Книга: Загадки сна
Назад: 7 Инсомния. Как победить бессонницу?
Дальше: 9 Храп и апноэ во сне. Храпеть не вредно?
8
Фармакология сна. Красная или синяя таблетка?
От дендрита – к синапсу. ГАМК – главная молекула сна. Победила ли валериана плацебо? Подарок святой Варвары. Лекарство против страха. Поколение Z. Почему аллергия мешает спать? Как обмануть внутренние часы? Мыши и болезнь Альцгеймера
Концепция управления сном базируется на модели регуляции сна, предложенной Клиффордом Сейпером в 2005 г. Согласно этой концепции, за контроль над функциональным состоянием мозга борются две системы – система поддержания бодрствования, так называемая активирующая, и система, вызывающая сон, – синхронизирующая (имеется в виду возникающая во сне синхронизация деятельности нейронов, которая выражается в замедлении ритма электрической активности). На стороне активирующей системы действует еще один «игрок» – внутренние часы (супрахиазменные ядра гипоталамуса). В светлое время суток эти ядра тормозят центр сна и усиливают активность центров бодрствования. С наступлением темноты тормозящее влияние уменьшается и центры сна берут верх, затормаживают центры бодрствования и заставляют нейроны коры разряжаться в пачечном, характерном для сна, режиме.
Напомним основы нейрофизиологии. Электрическое взаимодействие между нейронами в мозге осуществляется посредством синапсов (соединений). Типичный синапс обычно возникает между аксоном (длинным отростком) и дендритом (коротким отростком) нейрона. Между клеточными оболочками двух нейронов находится синаптическая щель – пространство, через которое от аксона к дендриту плывет химический передатчик информации – медиатор. Медиаторы, использующиеся в нервной системе, называются нейромедиаторами. Поверхности нейронов, обращенные внутрь синапса, содержат рецепторы, которые регулируют выделение медиатора (со стороны клетки – передатчика информации), рецепторы, реагирующие на медиатор (на противоположной стороне синапса), и рецепторы, влияющие на обратный захват клеткой-передатчиком уже использованного медиатора. Передача информации в синапсе возможна только в одну сторону – это направление от аксона к дендриту. Время, за которое медиатор преодолевает синаптическую щель, называется синаптической задержкой. Ее длительность составляет 0,5 миллисекунды. Соединяясь с рецептором на противоположной стороне синапса, медиатор вызывает в клетке-приемнике комплекс различных эффектов. В большинстве случаев эти эффекты сводятся к изменению электрического потенциала нейрона и передаче электрического импульса по цепи дальше. В одной клетке может образовываться несколько медиаторов, которые выделяются в одном синапсе, обычно один из медиаторов изменяет активность другого (является модулятором). Пример – нейропептид галанин, который вырабатывается в тех же нейронах, что и ГАМК, а именно – в гипоталамическом центре сна.
Основным веществом сна является гамма-аминомасляная кислота. Эта аминокислота является «универсальным тормозным медиатором» ЦНС. Соединяясь со специфическими рецепторами, ГАМК вызывает гиперполяризацию клетки (клеточная мембрана при этом приобретает еще более отрицательный заряд), что затрудняет генерацию потенциала действия (для этого клетке нужно достичь порогового значения в –55 мВ). Известно два типа рецепторов ГАМК. Наиболее распространены ионотропные ГАМКА-рецепторы. Они представляют собой ионные каналы в мембране нейрона, которые образованы пятью белковыми субъединицами. Известно 16 типов белков, образующих ГАМКА-рецептор: альфа (шесть изоформ – изоформа представляет собой любую из нескольких разных форм одного и того же белка), бета (три изоформы), гамма (три изоформы), а также дельта, эпсилон, пи и тета (одна изоформа в каждой). Главной из них является альфа-изоформа – соединение с ней вызывает комплекс реакций, зависящих от ее вида. Различные изоформы белковой альфа-субъединицы по-разному представлены в мозговых центрах, и в конечном итоге клинический эффект будет зависеть от того, какая именно изоформа альфа-субъединицы стимулируется. ГАМК стимулирует все изоформы – потому эта аминокислота и является универсальным тормозным медиатором. В дальнейшем были синтезированы химические вещества с избирательным действием на отдельные альфа-субъединицы, их клинический эффект является уже селективным в зависимости от того, в каком центре мозга больше представлена конкретная альфа-субъединица. Другие субъединицы, составляющие ГАМКА-рецепторный комплекс, имеют вспомогательное значение – они изменяют величину основного эффекта связывания ГАМК в большую или меньшую сторону.
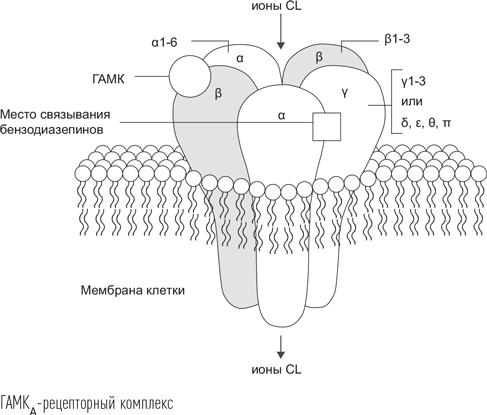
Каким же образом осуществляется передача сигнала в ГАМКА-рецепторе? Ионный канал – это отверстие в мембране клетки, через которое в клетку или из нее перемещаются ионы. В случае данного рецептора это ионы хлора, ток которых направлен внутрь клетки. Ионы хлора несут отрицательный заряд, поэтому чем больше их поступает в клетку, тем более отрицательным становится внутренний заряд мембраны. Поступление ионов хлора в клетку осуществляется за счет пассивной диффузии: поскольку в клетке хлора меньше, чем в межклеточной жидкости, то эти ионы стремятся проникнуть внутрь клетки. Пропускная способность хлорионного канала зависит от размера отверстия – чем оно больше, тем больше ионов поступит внутрь клетки в единицу времени. Присоединение ГАМК к центру связывания ГАМКА-рецептора вызывает изменение конформации белков, образующих отверстие; эти белки сворачиваются более плотно, и размер поры в мембране клетки увеличивается, что приводит к увеличению ионного тока внутрь клетки и к ее гиперполяризации.
Для того чтобы вызывать это изменение, ГАМК в рецепторе соединяется с определенной областью между двумя белковыми субъединицами – альфа- и бета-типов. Это место называют доменом связывания. Степень связывания ГАМК с рецептором в этом месте также зависит от активности других белковых субъединиц. Наиболее известна модуляция активности ГАМКА-рецептора при воздействии лекарственных средств группы бензодиазепинов (подробно об этом классе химических веществ будет рассказываться чуть позже). Их молекула связывается с другим доменом этого рецептора, находящимся между альфа- и гамма-субъединицами, что приводит к изменению конформации всего рецептора и повышает чувствительность домена, связывающего ГАМК. Другими словами, бензодиазепины повышают степень тормозного воздействия ГАМК на ГАМКА-рецептор.
Наличие в ГАМКА-рецепторе различных изоформ альфа-субъединиц и определяет клиническое действие этого нейромедиатора. Альфа-1 субъединица является самой распространенной – она входит в состав 60 % ГАМКА-рецепторов. Воздействие на рецепторы, содержащие именно эту изоформу альфа-белка, связывают с седативным и успокаивающим действием ГАМК, поскольку таких рецепторов достаточно много в интернейронах коры больших полушарий и ствола мозга. Также воздействие на эти рецепторы уменьшает вероятность эпилептического возбуждения (противоэпилептический эффект) и обеспечивает мышечное расслабление (миорелаксирующий эффект). Альфа-2 субъединица отвечает за специфическое анксиолитическое (буквально – «убирающее тревогу») действие: при применении веществ, избирательно связывающихся именно с этой субъединицей, отмечается уменьшение чувства тревоги и страха. Это объясняют тем, что альфа-2 субъединица встречается в ГАМКА-рецепторах миндалевидного тела (главного центра эмоционального реагирования) и коре больших полушарий. Анксиолитический эффект также наблюдается при активации рецепторов, содержащих альфа-3 субъединицу; они представлены в тех же областях мозга. Воздействием на ГАМКА-рецепторы, содержащие альфа-6 субъединицу, объясняется возникновение двигательных нарушений (неустойчивости при ходьбе), поскольку они в большом количестве присутствуют в мозжечке – главном центре координации двигательной активности и поддержания позы. За эффекты торможения процессов запоминания отвечает альфа-5 субъединица, большинство рецепторов, содержащих ее, находятся в гиппокампе – главном «процессоре» памяти.
Лекарственные препараты, основным действием которых является вызывание сна, называются снотворными. Наиболее часто главным механизмом, обеспечивающим снотворный эффект этих препаратов, является воздействие на ГАМКА-рецептор.
Снотворный эффект ГАМК обеспечивается воздействием на альфа-1 содержащие ГАМКА-рецепторы, находящиеся в главном центре сна – вентролатеральной преоптической области гипоталамуса (ВЛПО). Также было показано, что стимуляция этого же типа рецепторов в туберомамиллярной области гипоталамуса вызывает подавление активности одной из наиболее мощных активирующих систем – гистаминергической. Недавно было обнаружено, что снотворное действие ГАМК обеспечивается еще и воздействием на комплекс альфа-4-дельта субъединиц нейронов гиппокампа, таламуса и коры полушарий мозга. Таким образом, бытовавшее ранее представление о том, что снотворное действие препаратов, воздействующих на ГАМКА-рецептор, обеспечивается только усилением действия тормозных систем (центра сна), оказалось неверным. В этом процессе задействовано еще и подавление активирующих структур.
Сама по себе гамма-аминомасляная кислота снотворным не является, поскольку плохо проходит через гематоэнцефалический барьер. Это было обнаружено учеными в 1950-х гг., когда в Японии синтезировали первый аналог ГАМК – гаммалон. В дальнейшем предпринимались попытки повысить биодоступность, т. е. способность приникать в ЦНС, этого препарата путем присоединения к основной молекуле ГАМК различных радикалов (никотиновой кислоты, фенильного остатка), но эти средства также не обладали заметным снотворным действием.
Оказалось, что седативное и снотворное действие экстракта корня валерианы также объясняется воздействием на ГАМКА-рецепторный комплекс. Валериана является наиболее распространенным из безрецептурных лекарственных средств, используемых с этой целью. Она применяется как в чистом виде (в таблетках или настойках), так и в комбинации с другими лекарственными травами. Существуют препараты «Валокордин», «Корвалол», успокоительные сборы № 1 и 2, «Ново-Пассит». Имеется ли снотворный эффект у валерианы, или же ее действие обусловлено только эффектом плацебо? На этот вопрос попытались ответить ученые из Каталонского университета в 2010 г. Они собрали результаты всех исследований, в которых для улучшения сна применялись препараты валерианы и плацебо, и опубликовали метаанализ – статистически обработанный обзор этих исследований. Оказалось, что эффект валерианы не превышает эффекта плацебо в отношении сокращения времени засыпания или субъективно оцениваемого улучшения качества сна: при приеме валерианы время засыпания сокращалось по сравнению с плацебо на 0,7 минуты! Таким образом, принимая препараты, содержащие это вещество, чтобы уснуть, мы больше успокаиваем себя тем, что оно поможет, чем действительно получаем снотворный эффект. Следует отметить, что кроме воздействия на ГАМКА-рецепторы валериана, в отличие от других снотворных, действует еще на одни «сонные» рецепторы – рецепторы к аденозину 1-го типа (А1), однако, как мы видим, это не помогает вызывать сон – действие препаратов валерианы оказывается слишком слабым.
Первым агонистом (субстанцией, стимулирующей специфический рецептор) ГАМКА-рецепторов, полученным искусственно, стала барбитуровая кислота. Это химическое вещество было синтезировано в 1864 г. знаменитым химиком, нобелевским лауреатом Адольфом фон Баером, основателем химического концерна Bayer, которому принадлежит слава открытия аспирина (и героина). Согласно преданию, открытие барбитуровой кислоты произошло в день святой Варвары (4 декабря), отсюда и название этого вещества. Полученный фон Байером лекарственный препарат производился под названием «Веронал» – по названию города Верона в Италии. Он обладал успокаивающим и снотворным действием, предотвращал и погашал эпилептические припадки, поэтому получил широкое распространение в качестве замены распространенным тогда препаратам опиума как более эффективное и безопасное средство. К концу XX в. было синтезировано более 2500 химических соединений барбитуровой кислоты. Наиболее известным из них стало фенильное производное этой кислоты – фенобарбитал (люминал).
По современным представлениям, снотворное действие препаратов барбитуровой кислоты обусловлено связыванием с особой зоной ГАМКА-рецепторного комплекса, которая находится рядом с доменом связывания самой ГАМК. Этим, по-видимому, обусловлена широта клинического действия барбитуратов в связи со стимуляцией всех белковых альфа-субъединиц рецептора. Применение препарата сопровождается седативным, снотворным, противоэпилептическим, миорелаксирующим, амнестическим эффектами. Действие барбитуратов оказалось значительно сильнее эффекта самой ГАМК, поэтому они стали широко применяться в медицинской практике для лечения неврозов, нарушений сна, эпилепсии, болевых синдромов.
Когда первое восторженное впечатление от нового препарата улеглось, в ходе клинической практики стали проявляться проблемы, связанные с его применением. Оказалось, что фенобарбитал очень медленно перерабатывается и выводится организмом – период его полувыведения (время, когда в крови остается только половина концентрации) составил в среднем 79 часов – трое суток! Для каких-то медицинских целей, например для подавления тревоги или защиты от эпилептических припадков, это свойство было полезным. Однако для сна эффекты препарата оказались чрезмерными. Человек принимал фенобарбитал, чтобы выспаться: действительно, при этом он долго спал. Однако, вставая с постели утром, он ощущал сонливость, нарушение координации, затруднение мышления, поскольку действие препарата продолжалось. Электрофизиологические исследования показали, что сон на фоне приема препаратов барбитуровой кислоты становится неестественным – уменьшается количество самых восстанавливающих, глубоких стадий (3-я стадия медленного сна) и фазы быстрого сна. При этом после прекращения приема препарата наблюдался неприятный эффект «отдачи» – резкое увеличение количества этих стадий сна, что проявлялось множественными неприятными сновидениями и феноменом «сонного опьянения» (затруднением мышления после пробуждения).
В дальнейшем было обнаружено, что при длительном использовании препарата развиваются эффекты привыкания (требуется увеличение дозы для получения того же эффекта) и зависимости (при попытке отменить препарат состояние резко ухудшается – развивается так называемый «абстинентный синдром»). Было показано, что продолжительное применение фенобарбитала у детей приводит к задержке психического развития, ухудшению успеваемости в школе. Кроме того, в силу доступности и низкой стоимости препараты барбитуровой кислоты стали популярным средством сведения счетов с жизнью. Фенобарбитал оказался не очень удачным психотропным препаратом. Поэтому ученые продолжали работать над изучением клинических эффектов других агонистов ГАМКА-рецепторов, и к середине XX в. такое вещество было обнаружено.
Им оказалось производное бензодиазепина хлордиазепоксид, синтезированный в 1955 г. швейцарским химиком Лео Стернбахом. При испытаниях на животных у вещества обнаружились очень сильные седативный, противосудорожный и миорелаксирующий эффекты. Препарат получил название «Либриум» – «освобождающий». Он положил началу семейству бензодиазепиновых лекарственных средств. Бензодиазепинами эта группа была названа, поскольку химическая структура всех препаратов имеет в основании два кольца – шестичленное бензольное и семичленное с двумя атомами азота (диазепиновое). Изменять клинический эффект бензодиазепинов химики научились, присоединяя к этому основанию различные радикалы, а затем тестируя новое вещество на животных. Так появились диазепам, лоразепам, оксазепам, мидазолам и другие бензодиазепиновые препараты.
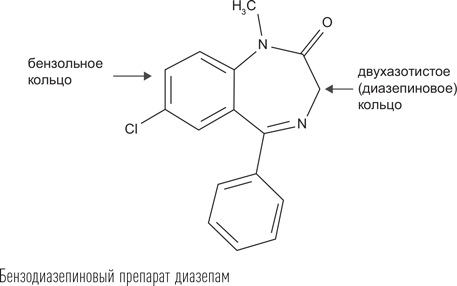
Они быстро вытеснили из клинической практики производные барбитуровой кислоты, поскольку, обладая таким же сильным действием, демонстрировали значительно более высокий профиль безопасности. Многие бензодиазепины имели короткий период полувыведения, значит, наутро они не вызывали сонливости, меньше действовали на координацию и когнитивные функции. Их эффект также был связан с воздействием на ГАМКА-рецептор, но объяснялся связыванием с особым доменом этого рецептора, который располагался между альфа- и гамма-субъединицами. Это приводило к усилению действия самой ГАМК, когда она соединялась со своим доменом. Было показано, что в отсутствие молекул ГАМК в синаптической щели бензодиазепины при соединении с рецептором эффекта не вызывали.
При использовании бензодиазепинов быстро выяснилось, что некоторые из них обладают большим снотворным действием, чем другие. Это свойство объяснялось сильным связыванием с альфа-1 субъединицей ГАМКА-рецептора. Поэтому препараты данной группы стали применять для получения снотворного и седативного (успокаивающего) эффекта. Другие препараты оказывали преимущественно анксиолитическое (противотревожное) действие, поскольку связывались в большей степени с альфа-2 и альфа-3 субъединицами. Их назвали «дневными» транквилизаторами, так как их седативный эффект был незначительным и позволял человеку активно функционировать днем. К этой группе относятся, например, медазепам и тофизопам. Бензодиазепины, обладающие выраженным миорелаксирующим или противоэпилептическим эффектом, также действуют на ГАМКА-рецепторы, содержащие альфа-1 субъединицу. К ним относятся диазепам и клоназепам.
В качестве снотворных бензодиазепиновые препараты прошли весь цикл испытаний, соответствующий требованиям доказательной медицины. Было проверено, действительно ли их эффект превышает действие плацебо. Для этого проводились рандомизированные двойные слепые контролируемые исследования. «Контролируемые» – это значит, что в качестве контроля эффекта выступала «пустышка», плацебо. «Слепым» называется исследование, когда больной не знает, получает он лекарственный препарат или плацебо, а «двойным слепым» – когда этого не знает и врач, поскольку пациенту назначается безликая, одинаково упакованная субстанция под номером, а номер выбирается случайным образом (этот процесс называется рандомизацией).
В этих исследованиях было показано, что на фоне приема бензодиазепиновых снотворных у людей с нарушением сна отмечается сокращение времени засыпания, увеличение общего времени сна, уменьшение числа пробуждений и времени бодрствования в ночное время. Преимущество снотворного эффекта по сравнению с эффектом плацебо было доказано для таких препаратов, как нитразепам, лоразепам, эстазолам, темазепам, триазолам, флюразепам (последние четыре в России не зарегистрированы). По сравнению с барбитуратами бензодиазепиновые снотворные оказались более безопасными – число побочных эффектов при их приеме в рекомендованных дозах было на порядок меньше.
Однако нежелательные побочные эффекты встречаются и при приеме бензодиазепинов. Эти препараты не были селективными и связывались не только с «нужной для сна» альфа-1 субъединицей. Параллельно они оказывали воздействие и на альфа-субъединицы второго, третьего, четвертого, пятого и шестого классов, вызывая кроме в общем-то полезного анксиолитического еще и нежелательные побочные эффекты. К ним относился феномен поведенческой токсичности из-за влияния этих препаратов на ГАМКА-рецепторы, расположенные в мозжечке и имеющие в своем составе альфа-1 и альфа-6 субъединицы. Наутро, когда препарат еще не полностью выведен из организма, у некоторых больных отмечалось нарушение равновесия. Встав с постели, такой человек мог внезапно пошатнуться и упасть. Поскольку нарушения сна чаще случаются у пожилых людей, массовый прием бензодиазепиновых снотворных препаратов привел к повышению частоты падений в этой возрастной группе, некоторые из таких происшествий заканчивались практически фатальным событием – переломом шейки бедра. После этого пожилой человек становился инвалидом, прикованным к постели, если ему вовремя не устанавливали протез тазобедренного сустава.
На фоне приема бензодиазепиновых снотворных кроме феномена поведенческой токсичности отмечалась еще когнитивная токсичность. Связывание препаратов с рецепторами, содержащими альфа-1 и альфа-5 субъединицы, которые широко представлены в гиппокампе и коре головного мозга, приводило к нарушению процессов внимания, мышления и запоминания. Получалось, что человек с нарушением сна принимает снотворный препарат, чтобы высыпаться и нормально работать, однако, несмотря на улучшившийся сон, возможности его деятельности оказывались снижены из-за действия снотворного препарата.
Другим нежелательным побочным действием бензодиазепинов так же, как и в случае с фенобарбиталом, оказалась проблема дневной сонливости, поскольку лишь немногие из бензодиазепиновых снотворных быстро выводились из организма. Самый распространенный в нашей стране бензодиазепиновый транквилизатор феназепам, например, имеет период полувыведения до 18 часов. Был обнаружен и другой потенциально опасный эффект бензодиазепиновых снотворных – усиление синдрома апноэ во сне. На этом состоянии мы подробно остановимся в следующей главе, пока же скажем, что применение таких снотворных приводило к увеличению числа остановок дыхания во сне (апноэ), что в еще большей степени ухудшало сон больных, вместо того чтобы его улучшить. Возникала парадоксальная ситуация: человек приходил к врачу, чтобы пожаловаться на то, что плохо спит, не зная, что частые ночные пробуждения у него связаны с реакцией организма на остановки дыхания. Врач, не проверив сон этого больного, назначал ему снотворный препарат, от которого число апноэ увеличивалось и пациент начинал спать еще хуже! Поэтому в инструкциях к большинству снотворных препаратов в настоящее время пишут «противопоказано» или «назначать с осторожностью» при подозрении на наличие синдрома апноэ во сне.
Сон на фоне приема бензодиазепиновых снотворных, увы, не становился идеальным. Несмотря на то что продолжительность его увеличивается и уменьшается число пробуждений, увеличение времени сна достигается за счет «среднего» по глубине периода сна (2-я стадия). При этом часто на фоне приема бензодиазепинов отмечается и подавление фазы быстрого сна. Когда же прием снотворного прекращают, то возникает «эффект отдачи» – резкое увеличение количества быстрого сна, как это случалось и при приеме барбитуратов. Таким образом, внедрение в практику бензодиазепиновых снотворных не вернуло страдающим бессонницей естественный восстанавливающий сон.
По мере накопления врачами опыта в условиях клинической практики оказалось, что прием бензодиазепинов, так же как и барбитуратов (хотя и в меньшей степени), может приводить к развитию привыкания и зависимости. В метаанализе исследований, включавших наблюдение за когнитивным статусом пациентов, принимавших бензодиазепины в течение многих лет, опубликованном в 2004 г., было показано, что их длительный прием сопровождается нарушением зрительно-пространственной памяти, снижением интеллекта, зрительно-моторной координации, процессов обработки информации, вербального обучения и концентрации внимания. Поскольку бензодиазепины стали доступными и относительно дешевыми психотропными средствами, за 10 лет (с 2005 до 2015 г.) число суицидов с их использованием увеличилось в три раза. Так что и эти снотворные препараты не стали избавлением от проблем, связанных с тревогой и нарушениями сна.
В 1986 г. «вышла в свет» новая группа снотворных – небензодиазепиновые агонисты ГАМКА-рецепторов. Первым из них стал зопиклон, одобренный к медицинскому применению британским здравоохранением. Затем появился золпидем, а последним в 1999 г. в США был одобрен к применению залеплон. Поскольку названия всех этих препаратов начинаются на Z, в дальнейшем их стали называть Z-препаратами. Особенностью этих лекарственных средств стало то, что, несмотря на отсутствие типичной бензодиазепиновой структуры (каждый препарат представлял собой уникальную молекулу), они селективно связывались с альфа-1 субъединицей ГАМКА-рецепторного комплекса, отвечающей за индукцию сна, и почти совсем не взаимодействовали с другими субъединицами. Поэтому их назвали первыми селективными снотворными.
Каждый из этих снотворных препаратов имеет особенности. Зопиклон – самое продолжительное время выведения из организма (время полувыведения – 5 часов) и небольшой дополнительный анксиолитический эффект. Поэтому его действия обычно хватало на всю ночь после однократного вечернего приема. Золпидем очень быстро всасывался в кровь и поступал в ЦНС, поэтому и действовать начинал очень быстро. Его период полувыведения оказался в два раза короче, чем у зопиклона, поэтому даже при приеме в середине ночи наутро нежелательный эффект «последействия» не отмечался. Последним из появившихся на рынке новых снотворных стал «ультракороткий» залеплон (период полувыведения 1 час), который так быстро удалялся из организма, что совершенно не ухудшал структуру сна, делая его практически естественным.
Многочисленные клинические исследования продемонстрировали, что по положительному эффекту на показатели сна Z-препараты оказались сравнимы с бензодиазепинами, а по вероятности возникновения побочных эффектов – значительно лучше их. Вероятность возникновения зависимости при приеме Z-препаратов оценивалась как в четыре раза меньшая, чем при приеме бензодиазепинов. Тем не менее возможность развития нежелательных побочных эффектов при приеме препаратов этой группы все равно существует, поэтому в инструкциях по медицинскому применению рекомендуется ограничиться 3–4-недельными курсами их приема. Более современные варианты Z-препаратов, такие как эсзопиклон, золпидем с медленным высвобождением, недоступные пока в России, можно применять в течение более длительного времени без риска развития зависимости, поскольку были проведены исследования, оценивавшие безопасность лечения этими препаратами в течение 6–12 месяцев.
Препаратом, действующим на другую разновидность ГАМК-рецептора – ГАМКБ-рецептор, является фенибут. Он представляет собой модификацию оригинальной молекулы ГАМК с целью обеспечения ей лучшего прохождения гематоэнцефалического барьера. Фенибут является отечественным изобретением – он был разработан классиком советской психофармакологии Изяславом Петровичем Лапиным в Ленинградском психоневрологическом институте им. В. М. Бехтерева и синтезирован в 1963 г. на кафедре органической химии Ленинградского педагогического института. Несмотря на отсутствие результатов плацебо-контролируемых исследований, этот препарат очень широко назначается врачами, особенно педиатрами, в качестве снотворного, ноотропного и мягкого успокаивающего средства (транквилизатора). Влияние фенибута на сон в сравнении с плацебо не изучалось, тем не менее в России его часто назначают в качестве снотворного средства.
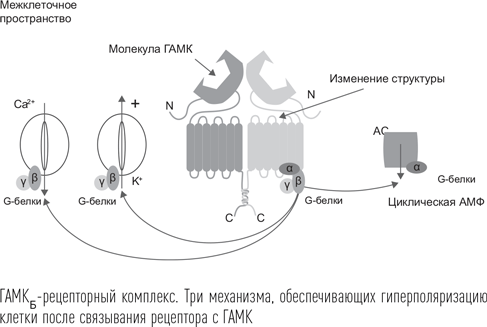
Особенностью функционирования рецептора ГАМКБ является то, что он действует по типу венериной мухоловки: когда молекула ГАМК подплывает к нему, одна из белковых субъединиц, составляющих рецептор, захватывает молекулу и блокирует ей выход, после чего запускается процесс изменения конформации белка, входящего в состав этого рецептора. ГАМКБ-рецептор относится к группе метаботропных рецепторов, связанных с G-белками. Соединение молекулы ГАМК с рецептором через эти белки вызывает каскад внутриклеточных процессов, результатом чего является увеличение выхода ионов калия (K+) из клетки, торможение входа положительно заряженных ионов кальция (Ca2+) и увеличение содержания циклической АМФ в клетке. Это в итоге приводит к возрастанию отрицательного потенциала клетки (гиперполяризации), и вероятность генерации ею потенциала действия снижается. Очень важной особенностью ГАМКБ-рецепторов является то, что большая их часть расположена не в области синаптической щели, а вне ее, на поверхности клеток. Поэтому они начинают реагировать, лишь когда выработка ГАМК очень сильно возрастает и этот нейромедиатор оказывается в межклеточном пространстве. Скорее всего, именно этим обусловлен довольно слабый психотропный эффект препаратов, действующих на этот рецептор.
Исследователи сна оценивают перспективы больных хронической инсомнией отказаться от снотворных довольно пессимистически. Показано, что до 70 % случаев инсомнии рецидивирует, т. е. через несколько лет болезнь возвращается. При этом до 40 % людей, которые отказались от приема снотворных, начинают принимать их вновь. Поэтому в настоящее время актуальной является задача поиска снотворного препарата (так называемое «идеальное снотворное»), который можно было бы принимать постоянно, каждую ночь, без риска развития привыкания, зависимости и снижения когнитивных функций в дальнейшем.
В связи с этим ведутся поиски других путей воздействия на качели сна, уже не со стороны усиления «сонных» влияний, а, наоборот, путем торможения активирующих систем. Один из этих путей – воздействие на нейроны, выделяющие гистамин.
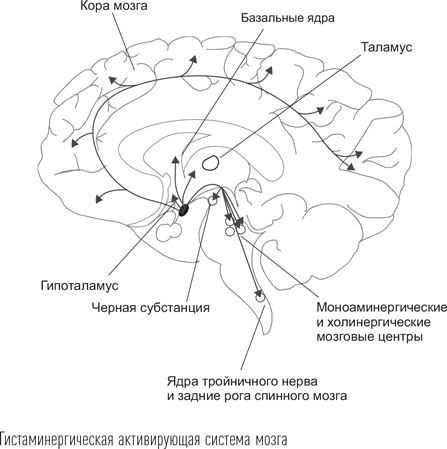
Гистаминергическая система головного мозга является одной из наиболее мощных активирующих систем несмотря на то, что содержит лишь небольшое количество нервных клеток (у человека – 64 000). В связи с большой степенью ветвления аксоны ее нейронов оканчиваются почти во всех важных мозговых центрах – базальных ядрах переднего мозга, таламусе, среднем мозге, черной субстанции, гиппокампе и миндалине. Благодаря этому гистаминовая система может стимулировать другие мозговые активирующие системы, например норадренергическую и ацетилхолинергическую. Особые отношения у мозгового гистамина с другим медиатором бодрствования – орексином. Орексиновая система активирует гистаминовую, но не наоборот. По образному выражению, «они формируют своего рода “оркестр”, в котором орексиновые нейроны играют роль дирижера, а гистаминовые – первой скрипки».
Снотворное действие вследствие торможения гистаминергической системы было известно давно, с момента изобретения первых антигистаминных средств. В 1907 г. британский ученый (в дальнейшем – нобелевский лауреат) сэр Генри Халетт Дейл открыл гистамин – один из основных медиаторов аллергических заболеваний. Это вещество секретируется тучными клетками соединительной ткани и базофилами (разновидностью лимфоцитов) крови. Выделение гистамина клетками вызывает системную реакцию, выражающуюся в повышении секреции слизи, затруднении дыхания, покраснении и зуде кожных покровов, головной боли, общем возбуждении, – эту системную реакцию называют аллергией. Крайний случай такой реакции, опасный для жизни, называется анафилактическим шоком, анафилаксией.
Первые химические препараты, блокирующие аллергическую реакцию, были синтезированы французскими учеными в Институте Пастера в Париже в 1937 г. Они разработали соединения, которые уменьшали выраженность анафилаксии у животных. Однако применение их у людей, больных аллергией, оказалось невозможным из-за высокой токсичности. В 1946 г. поступил в продажу под названием «Бенадрил» наиболее известный из всех антигистаминных препаратов дифенилгидрамин (в России он более известен как димедрол). Противоаллергическое действие этого лекарственного средства связывают с блокадой гистаминовых рецепторов 1-го типа (H1). Рецепторы данного типа присутствуют в гладких (непроизвольных) мышцах, клетках эндотелия сосудов и во многих центрах головного мозга.
Структурно H1-рецептор представляет собой так называемый метаботропный G-рецептор (рецептор, связанный с G-белками). G-группа – самая разнообразная и важная из всех рецепторов организма. Нарушение работы этих рецепторов приводит к возникновению множества различных заболеваний, а сами рецепторы являются мишенью для 40 % выпускаемых в мире лекарственных препаратов. Точный размер надсемейства рецепторов, связанных с G-белками, неизвестен, но почти 800 различных человеческих генов (или около 4 % от всего пула генов, кодирующих белки) были предсказаны из анализа последовательностей генома, кодирующих различные типы этих рецепторов. Семейство рецепторов, связанных с G-белками, включает рецепторы органов чувств (реагирующие, например, на свет или молекулы пахучих веществ): аденозина, бомбезина, брадикинина, эндотелина, ГАМКБ, меланокортинов, дофамина, адреналина, норадреналина, глутамата, ацетилхолина и серотонина, фолликулостимулирующего гормона, тиролиберина, окситоцина и многих других.
Поясним, каким образом активация G-рецептора приводит к изменению активности клетки. При соединении медиатора, в данном случае гистамина, с этим рецептором запускаются процессы, влияющие на скорость протекания химических реакций в клетке. Рецептором, связанным с G-белками, рецептор к гистамину был назван, поскольку состоит из семи белков, способных изменять активность гуанозиновых производных, присоединяя к ним молекулы фосфорной кислоты, и переводить эти молекулы в гуанозинтрифосфат (ГТФ). Когда сигнальная молекула (лиганд) связывается с частью G-белка, которая находится на поверхности клетки, то изменяется конформация другой его части, выходящей на внутреннюю поверхность клетки. В результате этот белок получает способность присоединить к связанной с ним молекуле гуанозиндифосфата (ГДФ) еще один остаток фосфорной кислоты, формируя энергетически насыщенную молекулу ГТФ. Эта молекула, будучи готовой, отщепляется вместе с частью G-белка (с его альфа-субъединицей) и уплывает, чтобы принять участие в химической реакции, а на ее место приходит другая молекула ГДФ. Система ГДФ-ГТФ является таким же поставщиком энергии для клетки, как и АМФ-АДФ-АТФ, но является более специализированной, принимая участие только в небольшом числе реакций.
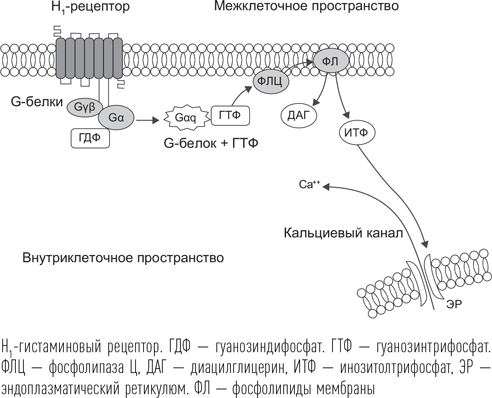
Получившийся комплекс ГТФ и альфа-субъединицы G-белка активирует вторичный передатчик (мессенджер) – фермент фосфолипазу Ц. Этот фермент «разрезает» молекулу фосфолипида так, что образуется диацилглицерол и инозитолтрифосфат. Инозитолтрифосфат внутри клетки связывается со своим рецептором на эндоплазматическом ретикулюме, вызывая высвобождение из этих закрытых хранилищ внутрь клетки ионов кальция (Ca2+). Накопление положительно заряженного кальция в клетке приводит к облегчению возникновения потенциала действия, поскольку клетка в итоге деполяризуется, отрицательный заряд на ее мембране становится ниже. Для мышечной клетки это приводит к сокращению, например, гладких мышц бронхов и затруднению дыхания при аллергии. Для чувствительного нервного окончания стимуляция рецепторов гистамином означает усиление импульсации, которую они посылают в головной мозг, что воспринимается как зуд или боль. И наконец, для нейронов, содержащих H1-рецептор, стимуляция гистамином выражается в усилении активности активируемых ими мозговых центров.
Итак, связывание гистамина или похожих с ним по структуре молекул со специфическим рецептором G-класса приводит к мозговой активации. Если вещество, связывающееся с H1-рецептором, будет похоже на него, но при этом не настолько активно, чтобы вызывать целевое воздействие (в данном случае – образование ГТФ), то этот рецептор перестанет работать, поскольку не сможет связываться с настоящим мессенджером – гистамином. Так работают блокаторы гистаминовых рецепторов. Центральное действие димедрола (дифенилгидрамина) связывают с еще более сложным феноменом – не блокадой, а «обратным агонизмом». Соединяясь с рецептором, молекула препарата «стабилизирует» его в неактивном состоянии, не давая возможности стимулировать клеточные химические процессы.
Довольно быстро выяснилось, что кроме желаемого противоаллергического действия блокаторы гистаминовых рецепторов вызывают целый ряд неприятных побочных эффектов – сонливость, сухость во рту, нарушение зрения, сердечную аритмию, снижение секреции желудочного сока. Последнее свойство связано с тем, что эти препараты блокируют другой тип гистаминовых рецепторов – H2, которые находятся в желудке и регулируют уровень секреции соляной кислоты. В дальнейшем для лечения язвенной болезни желудка были разработаны селективные блокаторы этих рецепторов. Другие побочные эффекты применения дифенилгидрамина, за исключением сонливости, оказались обусловлены блокирующим воздействием этого препарата не только на гистаминовые, но и на ацетилхолиновые рецепторы. При этом наблюдалось затруднение деятельности внутренних органов, управляемых парасимпатической частью нервной системы, переносчиком сигнала в которой служит ацетилхолин. При приеме этих препаратов развивались запоры, нарушался отток мочи и повышалось внутриглазное давление. С целью преодоления этих негативных эффектов в дальнейшем были разработаны селективные антигистаминные средства 2-го поколения, действующие только на периферические H1-рецепторы. Они не вызывают ни центральных, ни периферических нежелательных эффектов, поэтому для лечения аллергии применяют именно их. Примером такого препарата является антиаллергическое средство лоратадин (кларитин).
Снотворное действие, вызванное «неселективным» дифенилгидрамином, стало спасением для некоторых больных инсомнией, поскольку этот препарат оказался доступным и дешевым. Было проведено множество клинических исследований, подтвердивших, что дифенилгидрамин действительно улучшает ночной сон при его нарушении, в том числе и у детей. Поэтому перед учеными встала задача: «отделить» центральный снотворный эффект антигистаминного средства от периферического противоаллергического эффекта и синтезировать вещество, которое лучше подходило бы в качестве снотворного. Таким препаратом стал доксиламин. Первое упоминание о нем в медицинской литературе датируется 1948 г. Этот блокатор гистаминовых рецепторов не является полностью селективным, поскольку действует и на центральные, и на периферические рецепторы гистамина, на H1 и H2 их подтипы, а также на рецепторы к ацетилхолину. Но наибольшее сродство этот препарат проявляет к H1-рецепторам в головном мозге, при этом оказывается возможным подобрать дозировку, при которой будет достигаться снотворный эффект, в то время как вероятность развития нежелательных последствий окажется минимальной. Неожиданно обнаружилось, что доксиламин безопасен при беременности и в сочетании с витамином B6 может использоваться для лечения тошноты при этом состоянии.
Многолетний опыт использования блокаторов доксиламиновых рецепторов показал, что, хотя они действительно улучшают сон, их нельзя использовать в течение длительного времени из-за увеличения вероятности возникновения побочных эффектов. Это касается, прежде всего, развития дневной сонливости и снижения когнитивных функций, связанных с длительным периодом полувыведения препарата (около 15 часов). Кроме того, при применении препаратов этой группы существует риск развития привыкания – уменьшения эффекта лекарственного средства с течением времени. Поэтому в настоящее время дифенилгидрамин и доксиламин в качестве снотворных рекомендуется принимать лишь при острой инсомнии для того, чтобы помочь пациенту пережить период действия стрессового фактора, например социального конфликта.
Третий подход к фармакологической индукции сна заключается в использовании свойства внутренних часов (супрахиазменных ядер) влиять на взаимодействие активирующих и синхронизирующих систем мозга. Для этого используются препараты синтетического мелатонина – аналога гормона, вырабатываемого шишковидной железой (эпифиза). Раньше для этих целей использовался мелатонин, получаемый из мозга крупного рогатого скота, но в связи с потенциальной возможностью передачи через вещество мозга смертельного прионового заболевания (прионы – белки, обладающие способностью самокопироваться) – болезни Крейцфельда – Якоба – его стали производить генно-инженерным способом. Мелатонин, полученный таким путем, совершенно безопасен.
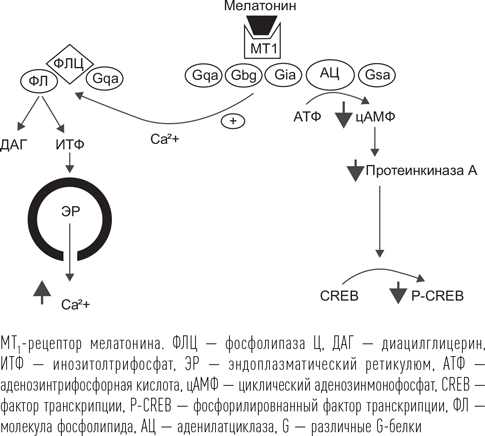
Препараты мелатонина воздействуют на специфические рецепторы 1-го и 2-го типа (МТ1 и МТ2). Рецепторы мелатонина также относятся к семейству рецепторов, связанных с G-белками, и, действуя через Gαi-белок, снижают уровень циклического аденозинмонофосфата (цАМФ). Гормон, взаимодействуя с рецептором, вызывает уменьшение образования цАМФ из АТФ, что, в свою очередь, активирует фермент протеинкиназу А, которая вызывает фосфорилирование (активацию) фактора транскрипции CREB. В итоге это приводит к изменению активности часовых генов. Также через систему инозинтрифосфата мелатонин вызывает увеличение содержания кальция (Ca2+) во внутриклеточном пространстве. Показано, что действие мелатонина на МТ1-рецепторы определяет индукцию сна у человека, а МТ2 отвечают за эффект подстройки собственного ритма внутренних часов к внешнему окружению (фазовый сдвиг).
Кроме МТ1 и МТ2 мелатонин действует на особые рецепторы, которые находятся внутри клетки, в ее ядре. Это рецептор RORα, участвующий в регуляции транскрипции циркадианных генов. Связывание мелатонина с этим рецептором напрямую вызывает изменение транскрипции часовых генов BMAL1, CLOCK, PER, CRY, CKIE, что в итоге приводит к изменению мембранного потенциала клетки в сторону его уменьшения. Клетке становится труднее генерировать потенциал действия. Поскольку 99 % рецепторов мелатонина находятся в головном мозге, точнее в супрахиазменных ядрах, основное действие этот гормон оказывает, затормаживая активность внутренних часов. В результате внутренние часы перестают «помогать» активирующим мозговым системам и тормозить синхронизирующие системы и равновесие между ними смещается в пользу сна.
Препараты мелатонина рекомендуется принимать в вечернее время, соответствующее началу его секреции у человека (в 21.00–22.00). Доказано, что прием мелатонина у здоровых людей сопровождается сокращением времени засыпания, а у людей с нарушениями сна – к сокращению времени засыпания и увеличению продолжительности сна.
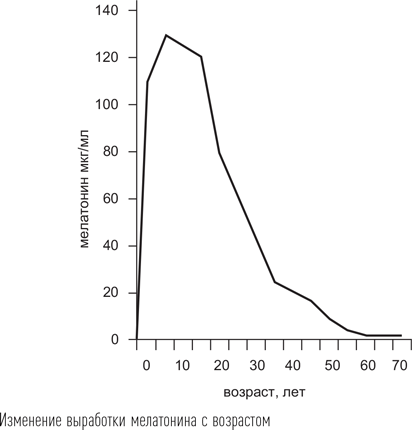
Особенностью мелатонина как лекарственного средства является его исключительная биодоступность – вследствие высокой жирорастворимости он легко проникает через мембраны клеток любого органа, включая головной мозг. Другая особенность – мелатонин очень быстро перерабатывается в печени: период его полувыведения не превышает 50 минут. Этим во многом обусловлена высокая безопасность препарата при приеме в качестве снотворного – наутро в организме не остается вещества, введенного извне. Показано, что препараты мелатонина становятся более эффективными в отношении улучшения сна по мере уменьшения его собственной продукции. В возрасте старше 55 лет собственный мелатонин почти не вырабатывается, поэтому в качестве снотворного его рекомендуется принимать, прежде всего, людям старших возрастных групп. Снотворный эффект препаратов мелатонина довольно слабый, слабее чем у любых других лекарственных препаратов, применяемых с этой целью.
Усилить действие мелатонина ученые пытались путем создания химически более активного вещества, которое связывалось бы с МТ1- и МТ2-рецепторами. При этом процессе ядро молекулы мелатонина сохраняется, но к нему добавляются активные радикалы, способствующие более тесной связи молекулы с рецептором. Поскольку структура G-рецептора досконально изучена, подбор подходящих молекул происходит методом компьютерного моделирования. Затем проводится тестирование лекарственных эффектов полученного вещества на животных. Новым агонистом мелатониновых рецепторов, используемым в качестве снотворного, стал рамелтеон, одобренный к применению в США в 2005 г. Это вещество сильнее, чем мелатонин, подавляет активность внутренних часов и способствует засыпанию. Его связывание с мелатониновыми рецепторами оказывается в 3–16 раз сильнее, чем у мелатонина. В клинических исследованиях было показано, что при длительном применении рамелтеона не развиваются привыкание и зависимость. В России этот препарат пока не зарегистрирован.
Эффект воздействия на МТ1- и МТ2-рецепторы используется и в новом антидепрессанте агомелатине. Это вещество связывается с МТ1-рецептором в два раза сильнее, чем мелатонин. При депрессии часто наблюдается нарушение различных биологических ритмов. В клинических исследованиях было продемонстрировано, что агомелатин не только улучшает настроение за счет блокады серотониновых рецепторов, но и способствует нормализации этих ритмов, в том числе и суточного цикла сон-бодрствование. Потому этот препарат рекомендуют больным депрессией с нарушением сна.
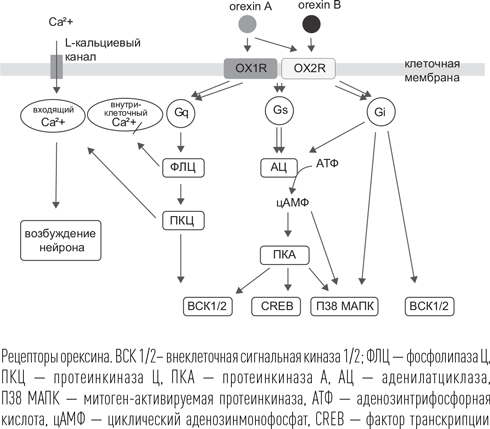
Наиболее «свежим» снотворным препаратом, появившимся на фармацевтическом рынке, является блокатор орексиновых рецепторов суворексант. В США он был одобрен к применению в 2014 г., в России – уже в 2016 г. Суворексант, подобно антигистаминным средствам, блокирует важную активирующую систему мозга, но на этот раз уже орексиновую, воздействуя на оба вида рецепторов орексина – 1-го и 2-го типов (OX1 и OX2). Эти рецепторы также относятся к семейству рецепторов, сопряженных с G-белком, их сигнальное действие осуществляется через механизм, подобный эффекту мелатонина. Соединение орексина с рецептором вызывает изменение конформации G-белков, что, в свою очередь, приводит к увеличению внутриклеточного содержания кальция через систему фосфолипазы Ц и протеинкиназы Ц (1-й путь); увеличению активности фактора транскрипции CREB, а также усилению активности киназ ВСК 1/2 и П38 МАПК (2-й и 3-й пути активации).
В плацебо-контролируемых исследованиях было показано, что применение суворексанта у больных инсомнией ускоряет засыпание, уменьшает число ночных пробуждений, удлиняет общее время сна и улучшает его эффективность. Это один из немногих снотворных препаратов, разрешенных к применению в течение длительного времени, поскольку суворексант показал свою эффективность и безопасность в течение года наблюдения. Интересно, что блокаторы орексиновой системы сейчас рассматриваются в качестве перспективных средств для замедления развития болезни Альцгеймера. В исследованиях, которые проводились пока только на мышах с искусственно вызванным заболеванием, применение другого блокатора орексиновых рецепторов, алморексанта, сопровождалось уменьшением выделения бета-амилоида и скорости образования бляшек – скоплений этого белка в головном мозге.
Итак, вы узнали, каким образом, опираясь на современную модель, базирующуюся на взаимодействии активирующих и синхронизирующих систем мозга, фармакологи управляют сном. Чаще всего для улучшения сна стараются усилить активность «сонных» систем, действуя на рецепторы ГАМК. Однако существуют и другие подходы. Можно блокировать некоторые активирующие системы, например гистаминовую или орексиновую. А еще можно «обмануть» внутренние часы, повлияв на мелатониновые рецепторы, и сместить таким образом равновесие двух систем в пользу сна. Когда врачи сталкиваются с необходимостью назначить пациенту с бессонницей снотворное, они выбирают препарат, основываясь на особенностях конкретного случая. При этом принимаются во внимание возможность получения дополнительного эффекта (например, подавление тревоги), наличие сопутствующих заболеваний (при повышении внутриглазного давления нельзя принимать блокаторы гистаминовых рецепторов), возраст пациента (у пожилых людей препараты мелатонина более эффективны) и продолжительность лечения (лишь немногие снотворные можно принимать в течение длительного времени).

