Книга: Пять литров красного. Что необходимо знать о крови, ее болезнях и лечении
Назад: Глава 23 Заболевания лимфатической системы. Кто такой Томас Ходжкин?
Дальше: Часть VI Лечение заболеваний крови
Глава 24
Хромосома родом из Филадельфии. Хронический миелолейкоз и другие миелопролиферативные болезни
В 1951 году Уильям Дамешек (1900‒1969) предложил объединить заболевания с трудновыговариваемыми названиями – полицитемию, хронический мегакариоцитарный лейкоз и идиопатический миелофиброз вместе с хроническим миелолейкозом – в группу хронических миелопролиферативных заболеваний. Во-первых, у них сходные клинические и морфологические свойства, а во-вторых, есть предположение, что они имеют общую природу происхождения.
Миелопролиферативные новообразования – название сложное, но давайте разберемся. Приставка myelo означает костный мозг, proles – потомство, ferre – нести = патологическое разрастание костного мозга в результате опухолевого процесса.
Дамешек назвал их «миелопролиферативные нарушения (disorders)», затем в 2008 году они были переименованы в «миелопролиферативные новообразования (neoplasms)». Это обусловлено открытием в 2005 году молекулярного маркера гена JAK2 киназы, общего для первичного миелофиброза, истинной полицитемии и эссенциальной тромбоцитемии. Тем самым была установлена взаимосвязь всех хронических миелопролиферативных лейкозов с мутационными изменениями в генах.
Миелопролиферативные новообразования (МПН) – группа заболеваний крови опухолевой природы, при которых в костном мозге вырабатывается слишком много клеток крови определенного типа. Частым общим для них клиническим проявлением является развитие очагов кроветворения вне костного мозга (чаще всего в селезенке) и тромбозов.
Важно, что миелопролиферативные новообразования – это хронические заболевания, которые требуют постоянной терапии и контроля.
Я не буду подробно описывать все заболевания этой группы, а остановлюсь на четырех самых распространенных: хронический миелолейкоз, истинная полицитемия, первичный миелофиброз и эссенциальная тромбоцитемия.
Хронический миелолейкоз
Хронический миелолейкоз (ХМЛ) – это клональное миелопролиферативное новообразование, возникающее в результате специфической хромосомной аномалии – образования той самой филадельфийской хромосомы, о которой мы говорили в главе 21. Помните историю про 9-ю и 22-ю хромосомы, которые обмениваются генами, «смешиваются» между собой? В результате образуется дефектный гибридный ген, который кодирует гиперактивную форму специфического белка – тирозинкиназы. Это и есть причина развития ХМЛ.
ХМЛ редкое заболевание, и частота его выявления составляет 1,3 случая на 100 тысяч населения в год, при этом мужчины болеют чаще женщин. Медиана возраста, когда заболевание диагностируется, – 59 лет.
Симптомы ХМЛ так же, как и при других гемобластозах, не специфичны: усталость, похудение, ночная потливость, температура непонятного происхождения, боль или чувство распирания под ребрами с левой стороны – там, где находится селезенка.
Как правило, эти симптомы развиваются тогда, когда масса накопленных опухолевых клеток начинает превышать 1 кг.
Для диагностики ХМЛ, как и при подозрении на любое гематологическое заболевание, необходимо выполнить полный клинический анализ крови. При ХМЛ в анализе крови обнаруживают высокое содержание лейкоцитов, иногда до очень высоких уровней. В крови появляются молодые (незрелые) формы – лейкозные бластные клетки и промиелоциты; большой процент зрелых клеток – миелоциты, метамиелоциты и нейтрофилы. В крови здоровых людей бластных клеток, промиелоцитов и миелоцитов не бывает. Появление незрелых форм лейкоцитов иногда наблюдается при тяжелых инфекциях, однако исчезает после лечения антибиотиками.
Помимо микроскопического исследования крови, при подозрении на ХМЛ выполняют анализ клеток костного мозга (миелограмму) и стандартное цитогенетическое исследование, его цель – найти ту самую филадельфийскую хромосому. Нет этой хромосомы – тогда пока воздерживаемся от диагноза «хронический миелолейкоз»: у больных до лечения она обнаруживается в 90‒95 % исследуемых клеток. Для цитогенетического исследования проводят аспирационную биопсию – берут жидкую составляющую костного мозга из костей таза.
Еще один важный анализ для подтверждения диагноза и оценки результатов терапии – молекулярный анализ. Он проводится из образца крови методом количественной полимеразной цепной реакции. При этом происходит подсчет количества сигнальных молекул BCR-ABL, которые ведут к усилению выработки опухолевых клеток.
После подтверждения диагноза «хронический миелолейкоз» определяют фазу заболевания. Различают три фазы:1. Хроническая фаза – это начальная стадия болезни, ее диагностируют у большинства пациентов – более 80 %.2. Фаза акселерации – ее определяют у 8–10 % первичных пациентов, и она представляет собой более продвинутую стадию.3. Бластный криз – наиболее агрессивная стадия ХМЛ, ее наблюдают в 1–2 % в дебюте заболевания.
Основная разница между фазами – это количество бластных (молодых, незрелых) клеток в костном мозге или крови.
Тактика терапии определяется индивидуально и зависит от большого количества факторов. Основные из них: возраст, фаза заболевания, общее состояние организма (наличие или отсутствие прочих хронических заболеваний), постоянная терапия другими лекарственными препаратами и т. д.
Самый главный тезис: абсолютных противопоказаний к назначению терапии не существует, то есть лечение возможно для всех, вне зависимости от возраста, хронических заболеваний и других факторов. Например, у меня есть пациентка, которая попала к нам в клинику в возрасте 85 лет, и на тот момент родственники уже готовы были с ней попрощаться. Да она и сама им говорила, что готова ложиться в гроб. Но не тут-то было!
Когда нам ее доставили, она была лежачая: слабость, потеря веса по неизвестной причине, практически не могла есть. Плюс куча различных заболеваний – диабет, гипертоническая болезнь и другие. Мы диагностировали у нее ХМЛ, выдали «волшебные» таблетки, которые были для нее наиболее безопасны, – и через месяц она вышла из клиники на своих ногах.
Через три месяца после выписки, когда она пришла на контрольный осмотр, мы ее не узнали: она вернулась к своему нормальному весу – и принесла каждому из нас по пирогу с капустой. Помню, отличные были пироги, с лавровым листом. На момент написания книги ей 93 года, она продолжает приходить на осмотры, и я регулярно получаю от нее приветы через коллег. Так что ХМЛ можно назвать излечимой болезнью практически в любом возрасте.
В наши дни применяется несколько способов лечения ХМЛ:
 таргетная терапия ингибиторами тирозинкиназ (ИТК) – стандарт лечения во всех фазах, самая эффективная терапия; существует уже три поколения этой группы препаратов;
таргетная терапия ингибиторами тирозинкиназ (ИТК) – стандарт лечения во всех фазах, самая эффективная терапия; существует уже три поколения этой группы препаратов; трансплантация костного мозга – проводится только при доказанной неэффективности первого способа;
трансплантация костного мозга – проводится только при доказанной неэффективности первого способа; терапия препаратами из группы интерферонов – до сих пор применяется как опция при неэффективности ИТК;
терапия препаратами из группы интерферонов – до сих пор применяется как опция при неэффективности ИТК; химиотерапия – применяется в некоторых случаях, но всегда в комбинации с ИТК.
химиотерапия – применяется в некоторых случаях, но всегда в комбинации с ИТК.Ингибиторы тирозинкиназ – самые эффективные препараты при ХМЛ. Благодаря им ХМЛ можно назвать одним из немногих онкологических заболеваний, для которого есть своя главная «волшебная» таблетка. Применение ингибиторов тирозинкиназ в течение последних 20 лет позволило значительно повысить эффективность лечения. Первым препаратом из этой группы был иматиниб, его открытие стало настоящим переворотом в лечении онкогематологических заболеваний.
Препарат «останавливает» развитие заболевания, то есть оно не прогрессирует до фазы акселерации и бластного криза в 92 %,.
При неэффективности или непереносимости иматиниба назначаются препараты следующих поколений,,.
Таким образом, вывод по поводу лечения ХМЛ звучит очень оптимистично: продолжительность жизни пациентов с ХМЛ ничем не отличается от длительности жизни условно здоровых – заболевание поддается лечению в подавляющем большинстве случаев.
Как оценивается эффективность терапии?
Конечно, было бы неплохо определить диагноз, выдать «волшебные» таблетки от ХМЛ и попрощаться, но пока это не так. После старта терапии начинается новый этап – наблюдение.
Основная задача любого лечения – максимальное снижение количества опухолевых клеток в организме и возврат к привычному образу жизни при максимальном сохранении ее качества.
В случае ХМЛ наша цель – получение и поддержание полного цитогенетического ответа. Мы должны добиться полного отсутствия клеток с измененной филадельфийской хромосомой.
По мере продолжения успешного лечения и достижения полного цитогенетического ответа число аномальных клеток будет прогрессивно снижаться, что свидетельствует о том, что заболевание оптимально поддается проводимой терапии.
Возможно ли полное излечение от ХМЛ? Устоявшийся тезис «терапия ингибиторами тирозинкиназ при ХМЛ – пожизненная и постоянная» в настоящее время все чаще подвергается сомнению.
После впечатляющих результатов терапии иматинибом ученые стали размышлять о возможности полного излечения и потенциальной вероятности отмены постоянной терапии у части пациентов.
Начиная с 2007 года проводятся клинические исследования по отмене лечения у пациентов. Первые данные были опубликованы в 2010 году группой французских ученых, а окончательные результаты увидели свет в 2017 году,. Другие аналогичные исследования продемонстрировали схожие результаты: чуть меньше половины пациентов могут находиться под наблюдением без терапии. В ближайшем обозримом будущем при идеальном мониторинге и достижении хороших результатов лечения часть пациентов будет обходиться совсем без терапии, а это, как вы понимаете, радикально скажется на их качестве жизни.
В моей практике есть много пациентов, которым была отменена терапия, – и у них все прекрасно. Особенно запомнился один, который очень боялся прекращать прием лекарств: «Я восемь лет принимал этот препарат и не знаю теперь, как без него жить». Действительно, у многих пациентов вся жизнь выстраивается вокруг болезни и ее лечения. И получается парадоксальная ситуация: я с радостью сообщаю человеку, что он излечился от злокачественного заболевания и может бросить пить таблетки, а он расстраивается. Отмена препарата вызывает тревогу: а вдруг болезнь вернется? Но с того дня прошло уже более семи лет. Пациент ходит к нам на регулярные осмотры и, кажется, привык жить без таблеток. Очень хочется верить, что в обозримом будущем эта опция будет доступна всем пациентам с ХМЛ.
Болезни без филадельфийской хромосомы
Теперь я расскажу вам о заболеваниях из группы Ph-негативных миелопролиферативных новообразований. Этот тип заболеваний называется Ph-негативным (читается как «фи-негативный»), потому что при нем, в отличие от хронического миелолейкоза, не обнаруживается филадельфийская хромосома – Ph-хромосома.
Ph-негативные миелопролиферативные новообразования по используемой сегодня в России Международной классификации болезней определяются как «новообразования неопределенного или неизвестного характера», то есть новообразования, вызывающие сомнения в том, являются ли они злокачественными или доброкачественными.
Получается, что пока у ученых нет единого мнения, к какой группе заболеваний их отнести – к доброкачественным или злокачественным. Но так как при этих болезнях значительно увеличивается риск развития тромботических осложнений и есть общие молекулярные поломки (мутации), а при длительном течении они способны трансформироваться в острые лейкозы, все-таки принято относить их к злокачественным.
Ph-негативные МПН схожи между собой повышенной частотой тромботических осложнений (закупорка сосуда тромбом). А как мы помним, сердечно-сосудистые осложнения (или тромбозы), например инфаркт сердца или инсульт головного мозга, остаются самой частой причиной смерти во всем мире.
При Ph-негативных МПН в крови значительно повышается количество всех клеток крови, и, когда их слишком много, они начинают закупоривать сосуды в организме. Тромбозы и тромбоэмболии сосудов различных органов и тканей – наиболее частое проявление этой группы болезней, именно они нередко становятся причиной обращения к врачам, что оканчивается установлением диагноза «опухоль крови».
Но есть и хорошие новости: все Ph-негативные МПН связаны с выявлением специфических мутаций, что сильно упрощает их диагностику. Для подтверждения диагноза Ph-негативных МПН обязательно выявление мутаций в генах JAK2, CALR или MPL.
Истинная полицитемия (ИП) – опухолевое заболевание крови, при котором в костном мозге образуется слишком много эритроцитов. Во многих случаях количество лейкоцитов и тромбоцитов также повышено. Проявляется ИП, как правило, разрастанием миелоидного ростка с возможным развитием внекостномозгового кроветворения (в селезенке), тромботическими осложнениями и вероятностью трансформации болезни в острый лейкоз.
Истинная полицитемия встречается с частотой 1,14 на 100 тысяч населения ежегодно. Женщины (1,28) болеют несколько чаще мужчин (0,99), со средней медианой возраста 65 лет.
Наиболее частые симптомы заболевания связаны с «переполнением» сосудов кровью, что влечет за собой расширение подкожных вен и покраснение кожи, повышение артериального давления, головную боль, головокружения, нарушение концентрации внимания. Из «странных» характерных симптомов – возникает кожный зуд (особенно после душа) и нестерпимые жгучие боли в кончиках пальцев рук и ног.
Диагностика заключается в проведении стандартных гематологических тестов: клинического анализа крови, в котором обнаруживается повышенное количество эритроцитов и, как следствие этого, повышение гемоглобина и гематокрита. Обязательно проводится аспирационная биопсия костного мозга с подсчетом миелограммы и цитогенетическим исследованием клеток костного мозга. Плюс необходимо молекулярно-генетическое исследование на наличие мутации JAK2. И последний, самый важный тест – трепанобиопсия костного мозга для гистологического исследования, при котором обнаруживаются характерные изменения.
Разработано много методов лечения ИП, контролирующих его течение путем снижения гематокрита до уровня ниже 45 %. Для удержания количества эритроцитов в пределах нормы очень важно, чтобы пациент находился под тщательным медицинским наблюдением и получал необходимое лечение.
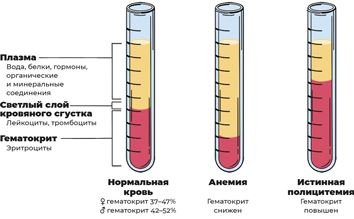
Рис. 26. Схематическое изображение процесса создания CAR-T-клеток
Несмотря на всю серьезность болезни, лечится оно «несерьезными» препаратами, знакомыми каждому. Например, для разжижения крови может назначаться аспирин в низких дозах. Для восстановления гематокрита – лечебные кровопускания (флеботомия). Также могут назначаться «циторедуктивные» препараты, снижающие количество клеток крови. Эта терапия будет пожизненной. Придется приспособить свое расписание не только под прием таблеток, но и под проведение регулярных кровопусканий: их необходимо делать в стационаре или поликлинике, но никак не дома. Кому-то достаточно проводить эту процедуру раз в два месяца, но есть пациенты, которым приходится ходить к врачу раз в две-три недели.
Эссенциальная тромбоцитемия (ЭТ) – заболевание, характеризующееся повышенным содержанием тромбоцитов в крови вследствие их чрезмерного образования в костном мозге из клеток-предшественниц, называемых мегакариоцитами. ЭТ регистрируется со средней частотой 1,18 на 100 тысяч населения каждый год, причем у мужчин чаще (1,46), чем у женщин (0,93). Медиана возраста заболевших – 67 лет.
Как уже говорилось, содержание тромбоцитов в крови здорового человека составляет 150×109‒400×109/л. Отклонение количества тромбоцитов от нормальных значений является одной из причин тромбоза и кровотечения. При ЭТ наблюдается стойкое повышение количества тромбоцитов выше 450×109/л.
Клиническая симптоматика эссенциальной тромбоцитемии может включать симптомы как тромбозов, так и кровотечений (частые носовые кровотечения, образование синяков без причины, кровь в каловых массах или в моче). Человек может регулярно сталкиваться с обмороками и предобморочными состояниями, а также испытывать жгучую или пульсирующую боль в кистях или ступнях, обусловленную нарушением кровотока (этот симптом называется «эритромелалгия»).
Диагностика эссенциальной тромбоцитемии ничем не отличается от диагностики истинной полицитемии, главное отличие – характерная гистологическая картина костного мозга, которую определяет врач-патоморфолог.
Терапия ЭТ также зависит от возраста пациента, наличия в прошлом перенесенного тромбоза и выявленной мутации. Основные цели терапии – предупреждение сердечно-сосудистых катастроф: тромбозов и кровотечений. Для этого применяются как выжидательная тактика, так и терапия препаратами, разжижающими кровь и снижающими количество тромбоцитов.
Первичный миелофиброз (ПМФ) – это миелопролиферативное новообразование, которое характеризуется фиброзом костного мозга, увеличением размеров селезенки и внекостномозговым кроветворением. Источником опухолевого роста является клональное расстройство гемопоэтической стволовой клетки.
Миелофиброз часто вызывает трудности при диагностике ввиду своей неспецифической клинической симптоматики. Например, хотя бы потому, что при нем выявляется три разные мутации с разной частотой: мутация гена JAK2 у 60 % пациентов, CALR – у 20–35 %, а мутация MPL – в 5–8 % случаев. Примерно у 10 % пациентов с МФ отсутствуют мутации перечисленных генов. В этом случае говорят о «трижды негативном» МФ, прогноз для которого обычно хуже.
 Миелофиброз – редкое заболевание и выявляется с частотой 0,33 на 100 тысяч населения ежегодно, мужчины болеют чаще (0,44), чем женщины (0,24). Медиана возраста на момент диагностики – 69 лет.
Миелофиброз – редкое заболевание и выявляется с частотой 0,33 на 100 тысяч населения ежегодно, мужчины болеют чаще (0,44), чем женщины (0,24). Медиана возраста на момент диагностики – 69 лет. Клиническая картина при миелофиброзе разнообразна и зависит во многом от стадии заболевания. При этом чаще всего встречаются следующие группы симптомов:
Клиническая картина при миелофиброзе разнообразна и зависит во многом от стадии заболевания. При этом чаще всего встречаются следующие группы симптомов:– повышенная утомляемость, слабость, одышка, бледность кожи, обычно обусловленные снижением количества эритроцитов, приводящим к анемии;
– боль в животе, ощущение тяжести или распирания в животе, снижение аппетита и потеря веса тела вследствие увеличения селезенки (спленомегалия);
– увеличение печени (гепатомегалия);
– кровотечения или кровоизлияния, возникающие беспричинно или после незначительных воздействий вследствие снижения количества тромбоцитов (тромбоцитопении);
– частые инфекции вследствие снижения количества лейкоцитов;
– стандартный набор малозаметных симптомов типа ночной потливости, зуда, лихорадки, боли в костях и суставах.
Миелофиброз грозен своими осложнениями, которые без должной терапии зачастую приводят к летальному исходу.
Главные из них – кровотечения, которые возникают из-за снижения числа тромбоцитов и по другим причинам; экстрамедуллярный (внекостномозговой) гемопоэз – ситуация, при которой костный мозг перестает производить клетки крови и вместо него это начинают делать другие органы, чаще всего – селезенка.
Это может стать причиной кровотечений из желудочно-кишечного тракта, кровохарканья или примеси крови в слюне, сдавления спинного мозга или появления судорог. Кроме того, из-за уплотнения костного мозга и воспаления соединительной ткани, окружающей кости, больной может испытывать сильные боли в костях и суставах. Эта боль может быть обусловлена также подагрой – одним из осложнений МФ. Ну и, кроме всего прочего, МФ может трансформироваться в острый миелоидный лейкоз (ОМЛ) – быстро прогрессирующее злокачественное заболевание крови (это происходит примерно у 15–20 % пациентов с МФ).
Диагностика ПМФ ничем не отличается от диагностики двух предыдущих заболеваний, и основными методами определения диагноза будут выявление характерных мутаций и гистологическое исследование костного мозга, без которого установить этот диагноз просто невозможно.
Лечение ПМФ состоит в сдерживании прогрессирования заболевания, улучшении качества жизни, контроле симптоматики и профилактике осложнений. Так как прогноз при этом заболевании неблагоприятный, следует на самом раннем этапе определиться с возможностью проведения аллогенной трансплантации костного мозга как единственно потенциально излечивающего метода лечения.
Миелопролиферативные новообразования – это группа хронических лейкозов, основная цель лечения которых – контроль над заболеванием и его осложнениями. В среднем продолжительность жизни пациентов при адекватной и своевременной терапии ничем не отличается от таковой в популяции.
Основные моментыМиелопролиферативные новообразования – это группа опухолевых заболеваний костного мозга, возникших в результате мутаций в кроветворной стволовой клетке и сопровождающихся бесконтрольным делением зрелых клеток крови.
Хронический миелолейкоз – хронический лейкоз, который развивается по причине специфической мутации – филадельфийской хромосомы, для лечения которой разработана высокоэффективная терапия – ингибиторы тирозинкиназ.
Классические Ph-негативные новообразования – это истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз. При этих заболеваниях выявляются специфические мутации генов JAK2, CALR и MPL. Для подтверждения диагноза обязательно требуется гистологическое исследование костного мозга. Основная цель лечения – предупреждение прогрессирования, контроль симптоматики и профилактика осложнений.
Тактика лечения Ph-негативных МПН основывается на следующих факторах: возраст, общее состояние организма, наличие тромботических осложнений в прошлом, тип выявленной мутации и возможность проведения трансплантации костного мозга.
Конечно же, злокачественных заболеваний крови и лимфатической системы гораздо больше, и они далеко не исчерпываются списком тех, что я привел. Хотелось бы мне остановиться подробно на каждом из них и рассказать про все методы сложной диагностики и принятие решений в непростых ситуациях, но поверьте мне на слово, их слишком много, чтобы втиснуть в эту книгу, а благодаря современным методам обследования становится еще больше. Я надеюсь, что совсем скоро будет доступна настоящая персонализированная терапия, основанная на полногеномном тестировании пациента, ведь каждый из нас уникален. Человеческая изобретательность безгранична, и я не сомневаюсь, что исследователи в скором будущем откроют высокоэффективные и безопасные методы лечения злокачественных заболеваний.
Назад: Глава 23 Заболевания лимфатической системы. Кто такой Томас Ходжкин?
Дальше: Часть VI Лечение заболеваний крови

