Болезнь Крейтцфельдта-Якоба и куру
В 1920 году немецкий нейрофизиолог Ганс Герхард Крейтцфельдт (правильнее – Кройтцфельдт) описал весьма необычное заболевание. 20 июня 1913 года к нему поступила пациентка Берта Е, у которой обнаруживались расстройства поведения и зрения, навязчивые мысли об одержимости, нарушение координации движений, эпилептические припадки. Болезнь развивалась два месяца – все это время больная находилась под наблюдением врача, но закончилось все летальным исходом 11 августа. Годом позже его соотечественник и коллега, невропатолог Альфонс Мария Якоб не просто описал симптоматику, но и связал ее с поражениями передних рогов спинного мозга и пирамидной системы – основных проводящих «магистралей» мозга, обеспечивающих движение.
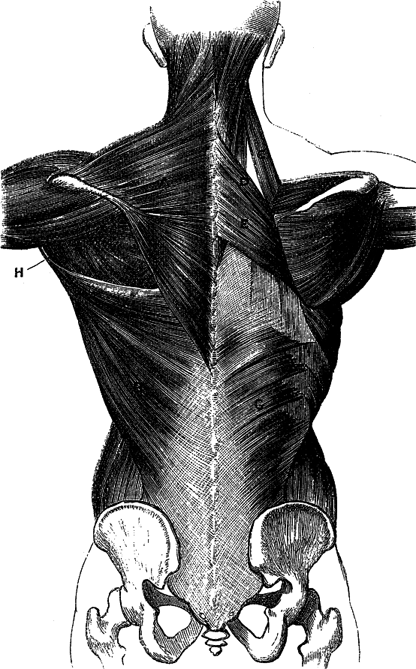
В самом мозге при этом происходили весьма интересные изменения. На гистологическом срезе ткани, особенно в мозжечке, исследователь замечал «губчатость», которая возникала из-за уже знакомой нам по мозгу овец вакуолизации нейронов, что создавало картину «пустот». Кроме того, он увидел еще одну деталь – странные белковые агрегаты, которые подобно мусору засоряют пространство между нейронами. Так мир узнал и о человеческой губчатой энцефалопатии, которую ранее уже описывали у овец и еще со Средневековья наблюдали среди крупного рогатого скота. Болезнь, как часто водится, назвали в честь первооткрывателей – болезнь Крейтцфельдта-Якоба (сокращенно БКЯ), хотя некоторые исследователи требуют пересмотреть название в сторону «болезни Якоба», поскольку только этот невролог описал саму патологию.
Поскольку понимание того, что подобные заболевания носят инфекционную природу, пришло только более чем 30 лет спустя и на примере с овцами, исследователи относили БКЯ к разряду энцефалопатий неясного происхождения (то есть «пока так, а потом разберемся»). До того момента, пока с юго-западной части Тихого океана не стали приходить любопытные новости.
В 1957 году американский врач словако-венгерского происхождения Даниел Карлтон Гайдушек (иногда пишут «Гайдузек» или «Гайдусек», но словацкая фамилия правильнее пишется именно так), педиатр по образованию, вирусолог по опыту и гений по природе, только что закончил работать в лаборатории сэра МакФарланда Бернетта в Австралии. На тот момент ему было всего 34 года, и его тщеславная натура стремилась реализовать свои амбиции. Он только что услышал от своего коллеги, что на островах Папуа Новая Гвинея, точнее – на одном из высокогорных склонов, где проживало племя форе, разыгралась лютая эпидемия, к тому моменту уже унесшая жизни 15 000 человек, из которых подавляющее большинство приходилось на женщин и маленьких детей. Для относительно небольшого племени это была настоящая катастрофа, поставившая его под угрозу вымирания.
С 1953 года среди них уже трудился Винсент Зигас, экспатриант из Прибалтики, ставший окружным австралийским врачом (тогда Папуа Новая Гвинея входила в состав Австралии). Ему-то на подмогу и отправили жаждущего приключений молодого детского инфекциониста, основная задача которого была лечить сифилис и дизентерию. Но он хотел большего – понять, что же за таинственное заболевание кроется под тем, что аборигены племени форе называли странным словом «куру».
«Куру» переводится как «тряска, дрожь» или «порча». Другое название болезни – «смеющаяся смерть» – целиком на совести газетчиков. Конечно, иногда она сопровождается дерганием головы и странной «сардонической» улыбкой, но все-таки главный симптом – дрожь, а потом уже и смерть – максимум через год.
Распространялась болезнь нетривиальным способом – через ритуальный каннибализм. У этого племени было принято съедать своих родственников прямо в ходе ритуала погребения – считалось, что таким образом они навсегда остаются с живыми и передают им свои знания и опыт. Мясо, как правило, доставалось мужчинам, а вот женщинам и детям оставляли внутренности и самое главное – мозг, для «мозговитости» и развития ума. Так человек ел мозг инфицированного или заболевшего куру (не специально выбирая, конечно) и заражался сам. Отсюда и специфичный гендерный перекос.
Гайдушек никак не мог понять, с чем имеет дело. Он наблюдал маленьких истощенных детей, которых одолевала дрожь вплоть до того, что они не лишались сил и не падали наземь окончательно. Когда они умирали, он, иногда получая разрешение их вскрывать и даже брать образцы крови, внутренних органов и мозга, тщательно собирал все вещественные доказательства и отправлял в США. Там ему помогал Игорь Клатцо, невропатолог из Национального института нервных болезней США, обладавший всем необходимым для решения технической стороны вопроса.
Именно ему бросилось в глаза сходство мозжечка умершей от куру представительницы форе с картиной мозга, пораженного болезнью Крейтцфельдта-Якоба, которую он когда-то видел в коллекции своего учителя, немецкого невролога Оскара Фогта. Гистологические препараты получились настолько необычными (кто ж останется равнодушным, созерцая дыры в мозге, подобные срезу хлебной буханки), что Клатцо решил направить их на ежегодную медицинскую выставку в Лондон.
Почти сразу после окончания выставки доктор Клатцо получил сообщение от английского ветеринара Билла Хадлоу, который указал на сходство мозга овец при скрепи и увиденных им препаратов. Чтобы разгадать тайну куру, он порекомендовал исследователям попытаться заразить обезьян как наиболее близких к человеку млекопитающих, которые теоретически могут дать нужную реакцию для подтверждения факта инфицирования.
Сказано – сделано. Гайдушек выпросил у местных жителей порцию мозга очередного почившего больного и накормил кашицей из него пятерых шимпанзе, а также некоторую другую живность – мышей, морских свинок, крыс и даже цыплят. Он ждал результата два месяца, полгода, год – Хадлоу предупредил о долгом инкубационном периоде – но так ничего и не дождался. Ни одного намека на дрожь и истощение. Неужели это генетический недуг?
Однако начиная с 1959 года власти Австралии ввели запрет на каннибализм и начали упорно его искоренять, жестко карая непослушание, и с этого времени болезнь внезапно пошла на спад. Хотя, конечно, отдельные вспышки потом наблюдались еще в течение 30 лет. В порыве откровенности аборигены признавались, что тайком продолжали лакомиться мозгом усопших, но начиная с 60-х годов случаи поедания себе подобных прекратились вовсе. Так же, как и прекратили болеть маленькие дети.
Все эти факты, а также наблюдения за ритуальными церемониями сложились в голове у Гайдушека в единую картину – несет болезнь не сам факт поедания пораженного мозга, а то, что женщины свои руки, которыми этот мозг измельчают и раскладывают по порциям, не моют. Патогенные частицы с них могут попадать куда угодно – в царапины, в глаза, укусы и расчесы. Логическая цепочка замкнулась.
Недолго думая, исследователь снова организовал эксперимент с шимпанзе. Теперь смертельная мозговая кашица вводилась животным прямо под кости черепа. Оставалось лишь дождаться…
И ожидания оправдались – через 21 месяц (!) у одной из обезьян появились первые симптомы, и уже ее мозжечок послужил смертельным эликсиром для следующего поколения подопытных животных. Череда успешных испытаний завершилась лишь к 1966 году, зато после этого ни у кого уже не осталось сомнений об инфекционной природе заболевания. И, судя по всему, близкой к скрепи или БКЯ. Работа, достойная публикации в Nature.
Гайдушеку удалось экспериментально установить инфекционный характер заболевания, однако он сам придерживался «основной линии партии» и считал, что куру, как и БКЯ, инфекционную природу которой установил он же, вызывается некими «медленными вирусами». Ведь он был вирусологом. Тем не менее в 1976 году именно за это открытие он получил Нобелевскую премию (вместе с исследователем гепатита В Барухом Бламбергом). Кстати, денежную часть премии Гайдушек пожертвовал племени форе.
В те же 60-е годы следующий и невероятно важный шаг к пониманию причин болезней сделали британцы – радиобиолог Тивах Альпер и математик Джон Стенли Гриффит. Они независимо друг от друга пророчески предположили, что загадочный болезнетворный агент слишком мал для вирусов – уж очень велика доза ионизирующего излучения, необходимая для того, чтобы уничтожить половину инфекционных частиц (чем меньше размер объекта, тем меньше вероятность попадания в него заряженной частицы, значит, нужно больше частиц). Вывод напрашивался сам собой: если это не вирусы, то… белки?
Кстати, потом эту теорию высоко оценил Френсис Крик. Но поначалу со всех сторон на исследователей посыпались возгласы непонимания, смешки и сарказм. И вот тут настал черед выхода на сцену очередного героя инфектологии – Стэнли Прузинера.
Нужно сказать, что Стэнли – очередной нобелевский лауреат с российскими корнями. В 1896 году его дед по отцовской линии, Беня Прузинер (Пружинер), эмигрировал в США из Москвы. Как писал сам ученый в автобиографии, опубликованной на сайте Нобелевского комитета, его жизнь нетипична для американца: родился на Среднем Западе, учился на Востоке, а живет на классическом Западе. Свое имя он получил в честь брата отца, умершего в возрасте 24 лет от лимфомы Ходжкина. Его отец был военным моряком, прошедшим Вторую мировую войну и потом участвовавшим в испытании первой американской водородной бомбы.
Средняя школа Walnut Hills запомнилась Прузинеру исключительно пятилетним курсом латыни (которая, по его словам, потом сильно помогла писать научные статьи), а так показалась скучной. Бакалавриат Пенсильванского университета Прузинер закончил по химии, но потом остался на местном медицинском факультете, где изучал сначала гипотермию, а затем – флуоресценцию жировой ткани сирийских хомячков(!).
Интернатуру он проходил уже в Калифорнийском университете, параллельно продолжая карьеру в знаменитом Национальном институте здоровья (NIH), где изучал ферменты глутаминазы бактерии E. coli под руководством Эрла Стадтмана. Устраиваясь в институт, он долго колебался – сможет ли? Однако привилегии, которые давал NIH, перевесили. Результат трех лет работы сам Прузинер описывает коротко: выжил.
Впервые исследователь столкнулся с якобы «медленным вирусом» в 1972 году, когда приступил к работе в отделении неврологии Калифорнийского университета (Сан-Франциско). Всего через два месяца после начала работы у него умерла пациентка от необратимых повреждений мозга, вызванных болезнью Крейтцфельдта-Якоба. Именно тогда он узнал, что ученые до сих пор не уверены, что эту болезнь вызывают именно вирусы. И честолюбивый молодой врач решил, что открытие молекулярной структуры возбудителя CJD (такой английской аббревиатурой обозначается в литературе БКЯ) станет хорошим началом самостоятельной научной карьеры.
Два года работы с литературой позволили Прузинеру понять, что это будет непросто. Тем не менее, он открыл свою лабораторию в 1974 году, хотя для поддержки работ по изучению БКЯ ему пришлось писать заявки на гранты по глутаматному метаболизму. «Скучно, но у меня был опыт», – пишет Прузинер.
В итоге первая статья о выделении нового агента – белка-приона – вышла только в 1982 году. И именно Прузинер стал автором термина «прион». Это название происходит от «склейки» двух английских слов: proteinaceous infection (белковая инфекция). Статья вызвала настоящую бурю, еще бо́льшую, чем была после объявления о догадках, которые сделали Альпер и Гриффит – медицина вообще консервативна, но предположить, чтобы заболевания передавались белками?! Далеко не все приняли эту концепцию. Собственно говоря, Гайдушек так и не признал открытие Прузинера вплоть до своей смерти в 2008 году.
Десять лет спустя (то есть в 1992 году) Прузинер выпустил солидный итоговый труд – «Молекулярную биологию прионных болезней». Ну а еще через пять лет пришла долгожданная и очень желанная награда. За раскрытие тайны прионов Стэнли Прузинер в 1997 году удостоился Нобелевской премии, а возбудитель болезни – упоминания в формулировке Нобелевского комитета: «За открытие прионов, нового биологического принципа инфекции».
«Люди часто спрашивают меня, почему я упорствовал в исследовании столь спорного предмета. Я обычно отвечаю, что всего лишь нескольким ученым выпала великая удача изучать темы, столь новые и необычные, что только небольшое число людей может осознать значение таких открытий с самого начала. Я – один из тех по-настоящему везучих ученых, которому представилась особая возможность работать над такой проблемой – проблемой прионов», – говорил на своей Нобелевской лекции Прузинер.
Прионы, как выяснилось – это не новая форма жизни, а собственные белки человека, ставшие патогенными из-за изменения своего пространственного строения, конформации. Оно может быть вызвано разными причинами – от внешних воздействий до генетических изменений. В своей неинфекционной форме прионы входят в состав нормальной нервной ткани. Но как только в организм попадают прионы в инфекционной форме, они резко увеличивают свою численность, придавая инфекционную конформацию своим ранее безобидным копиям, имеющимся в нейронах. В результате в мозге постепенно накапливается огромное количество нефункционального белка, делающее невозможной работу нервных клеток. Из-за их гибели в мозге и появляются характерные «дыры».
Куру так и осталась очень ограниченным и обособленным «уделом» племени форе, а вот скрепи, коровье бешенство и болезнь Крейтцфельдта-Якоба, встречаемая у людей, есть и сейчас. Выделяют несколько ее форм, различающихся происхождением: классическую (возникает спонтанно, 85 % всех случаев БКЯ), наследственную (возникают мутации в гене PRNP человеческого прионного белка, 10–15 % случаев) и новый вариант (то самое коровье бешенство, которым заражаются при поедании прионсодержащей говядины). Ранее, когда еще не знали о происхождении БКЯ, встречались ее ятрогенные варианты, когда патогенные прионы распространяли в процессе медицинских манипуляций или в составе препаратов из тканей и биологических жидкостей животных/человека.
Несмотря на то, что сейчас мы знаем о прионных заболеваниях достаточно много, разобрались в их молекулярном происхождении и поняли, как можно обезопаситься хотя бы от тех форм, которые поддаются профилактике, лечить их мы не научились. Более того – сейчас пришли к мнению, что прионы способны образовывать в мозге бляшки, подобные тем, которые встречаются при болезни Альцгеймера.
Совсем недавно стало ясно, что прионы также вызывают пигментный ретинит – дегенеративное заболевание сетчатки, которое, как считалось, вызывается проникновением в глаз клеток микроглии, которые разрушают фоторецепторные клетки. Исследователям удалось выяснить, что интенсивное накопление молекул прионов вблизи сетчатки, происходившее одновременно с ее прогрессирующим разрушением, свидетельствует о том, что пигментный ретинит – это еще одно прионное заболевание.
Появились и идеи насчет того, как прионные заболевания лечить.
В августе 2019 года стало известно, что исследователи из США смогли замедлить течение прионных болезней у модельных животных. Очередная попытка использовать для этого антисмысловые олигонуклеотиды на сей раз увенчалась успехом. Работа, сулящая прорыв в терапии, была опубликована в журнале JCI Insight.
Идея антисмысловой терапии заключается в следующем. Как известно, синтез белка происходит по схеме: информация об аминокислотной последовательности закодирована в гене. Она считывается РНК, после чего на матрице РНК в рибосоме синтезируется белок. Если ввести в организм так называемые антисмысловые нуклеотиды, то они свяжутся с матричной РНК и не позволят рибосоме считать с нее структуру белка: трансляция адресного гена блокируется. В итоге нужный нам белок не синтезируется.
Сотрудники Национального института аллергии и инфекционных заболеваний и Института Броуда использовали подобные олигонуклеотиды в борьбе с мышиной моделью скрепи (почесуха овец). В этом случае мыши сначала заражались непосредственной инъекцией препарата гомогенизированного мозга мышей со скрепи в терминальной стадии, а затем им вводили профилактическую инъекцию антисмысловых олигонуклеотидов (ASO) к гену прионного белка Prnp непосредственно в желудочки мозга.
Предыдущие попытки использовать антисмысловую терапию проводились постоянным введением небольших доз и не принесли особого успеха. На этот раз авторы исследовали два других метода. В первом мышам делалась однократная инъекция большой дозы препарата примерно на 120-й день жизни – незадолго до обычного времени первого появления симптомов у мышей линии скрепи. В этом случае появление симптомов удалось отодвинуть на 55 процентов (87 дней). Во втором случае мышам делались инъекции каждые 2–3 месяца. Как отмечают авторы, в этом случае продолжительность жизни экспериментальных животных увеличивалась на 61–98 процентов.
Если учесть, что прионные заболевания, как правило, вообще неизлечимы, это – очень хороший результат. Особую важность он обретает в свете совсем недавней работы, показывающей прионный характер распространения болезни Паркинсона из кишечника по блуждающему нерву в мозг.
Дело в том, что в июне 2019 года исследователи из Института клеточной инженерии Университета Джонса Хопкинса опубликовали в журнале Neuron, одном из топовых изданий в области нейробиологии, статью, в которой они экспериментально показали, что болезнь Паркинсона может зарождаться в кишечнике и затем по блуждающему нерву постепенно проникать в мозг.
В своей работе авторы проверяли гипотезу, которую высказал в 2003 и 2004 годах Хейко Браак. Изучая патологию болезни Паркинсона, он обнаружил на ранних стадиях тельца Леви, в которых концентрируются альфа-синуклеиновые фибриллы в обонятельной луковице, а также в дорсальном моторном ядре блуждающего нерва (DMV) продолговатого мозга. Посему Браак предложил гипотезу о том, что болезнь Паркинсона начинается в кишечнике, а затем «перебирается» по блуждающему нерву в мозг, где и разрушает дофаминергические нейроны черной субстанции.
Кроме того, было известно, что фибриллы альфа-синуклеина, состоящие из неправильно свернутых молекул, могут «заражать» здоровые белки и передаваться от клетки к клетке наподобие прионных заболеваний. Ну и работы 2009 и 2012 года показали, что рекомбинантно синтезированный альфа-синуклеин способен «собираться» в фибриллы (PFF), которые распространяются как в культурах нейронов, так и в живом мозге.
Наконец, «неправильный» альфа-синуклеин можно обнаружить по фосфорилированному серину (специфическому биохимическому показателю) в 129-м аминокислотном остатке.
Итак, что же сделали авторы работы? Они взяли мышей и ввели им PFF в мышцы, обволакивающие двенадцатиперстную кишку. Эти мышцы обильно иннервируют ответвления блуждающего нерва. И за ними начали следить.
Через месяц фосфорилированный серин обнаружился в альфа-синуклеине в продолговатом мозге. Через три – в среднем мозге и миндалевидном теле, через семь – в переднем мозге и гиппокампе. Параллельно начались симптомы болезни Паркинсона.
Чтобы убедиться, что распространение происходило именно через вагус (так еще называется блуждающий нерв), другую порцию несчастных экспериментальных животных подвергли вагэктомии – пересечению блуждающего нерва. Мозг прекратил «общаться» с кишечником, у мышей наблюдались проблемы с аппетитом – но зато распространения белка-убийцы не происходило. Для верности исследователи взяли мышей, нокаутных по белку альфа-синуклеину (Snca−/−). Распространения патологической формы тоже зарегистрировать не удалось, и это показывает, что молекулы по нерву не путешествуют, а лишь «заражают» здоровые белки, распространяясь по прионно-подобному механизму.
Так что возможно, прионные болезни распространены несколько серьезнее, чем загадочная болезнь куру. Но, возможно, мы уже подбираемся к их лечению.
Ну и напоследок – очень интересный факт. Ровно через год после смерти Карла Гайдушека, первооткрывателя болезни куру, нам стало известно, что некоторые члены племени форе, благодаря появившемуся у них в сравнительно недавнем времени новому полиморфизму гена PRNP, имеют врождённый иммунитет к куру. Эволюция в действии!

