Книга: ДНК. История генетической революции
Назад: Глава 11 Генетическая дактилоскопия. ДНК в суде
Дальше: Глава 13 Что важнее: наследственность или воспитание?
Глава 12
Гены и болезни: поиски и лечение
Было настолько раннее и безлюдное утро, что даже встреча с пьяным человеком в это время казалась странной. Еще более удивительно было увидеть в этот час пьяную, немолодую, нелепо одетую женщину. Она шла по улице виляющей из стороны в сторону походкой, и всем, кто ее тогда видел, она казалась именно пьяной, даже постовому, дежурившему рядом со зданием суда, который сделал ей замечание, чтобы та в свои годы не позорилась. На самом деле Леонора Векслер не была пьяна. Ее начинал одолевать злой рок, который уже свел в могилу нескольких ее родственников прямо у нее на глазах – участь, которая, как она надеялась, ее минует.
Вскоре после описываемого эпизода, в 1968 году, Милтон Векслер, бывший муж Леоноры Векслер, должен был отпраздновать в Лос-Анджелесе свое шестидесятилетие вместе с двумя дочерями: Элис (ей было 26 лет) и Нэнси (23 года). Однако, как оказалось, семья собралась вместе не только ради торжества. Милтон сообщил дочерям, что их пятидесятитрехлетняя мать страдает болезнью Хантингтона, разрушительным неврологическим расстройством, вызывающим прогрессирующую деградацию функций мозга: больные постепенно забывают абсолютно все и о близких, и о себе самих. Они также теряют контроль над собственными конечностями. Сначала нарушается походка, как в случае Леоноры, но по мере нарастания деградации больной также начинает совершать непроизвольные конвульсивные движения. Выражаясь научной терминологией, болезнь Хантингтона – наследственное заболевание, при котором у людей среднего возраста появляются периодические мышечные подергивания или спазмы и происходит постепенная дегенерация мозговых клеток, приводя к хорее, атетозу и ухудшению интеллектуальных функций. Лекарств, способных отсрочить это неуклонное движение к смерти, не было.
Элис и Нэнси вспомнили, что трое их дядюшек, братьев Леоноры, умерли в молодости, как и отец самой Леоноры, Абрахам Сейбин. Они уже понимали, что болезнь Хантингтона – наследственная. Милтону выпала тяжелая доля ответить на их естественный вопрос: «А какова вероятностьтого, что то же самое случится с каждой из нас?» Ответ отца был: «Пятьдесят на пятьдесят».
Болезнь, о которой мы говорили выше, открыл Джордж Хантингтон. Сам он родился в семье врачей и вырос в Ист-Хэмптоне на Лонг-Айленде, где еще мальчиком сопровождал отца на обходах больных. Получив диплом врача в Колумбийском университете, Хантингтон на несколько лет вернулся к семейной практике на Лонг-Айленде, а затем перебрался в Помрой, штат Огайо. В 1872 году он представил в Медицинской академии Мейджса и Мейсона в расположенном неподалеку Миддлпорте свою статью, озаглавленную «О хорее». Термин «хорея» происходит от греческого слова, означающего «танец»; с XVII века врачи называли этим словом болезни, при которых у больных наблюдались конвульсивные движения.
В статье молодого врача был мастерски описан синдром, позже получивший название «хорея Хантингтона», а ныне известный как «болезнь Хантингтона». В статье признавалось, что эта болезнь наследственная: «Если болезнь проявилась у одного или у обоих родителей, то один или более их отпрысков также неизбежно заболеют. Болезнь не минует ни одного поколения. Вступив в свои права, она остается у человека на всю жизнь». Иными словами, болезнь Хантингтона является наследуемой по доминантному типу. Также было очевидно, что она в равной степени поражает как мужчин, так и женщин, то есть ее ген находится где-то в двадцати двух неполовых хромосомах (аутосомах).
Тогда, в 1968 году, о болезни Хантингтона было известно не так много: она наследственная, необратимо прогрессирует, убивая нервные клетки в конкретных зонах мозга. Милтон Векслер основал Фонд по борьбе с наследственными заболеваниями (HDF) для сбора денег на исследование болезни Хантингтона и привлечения к ней внимания властей. Его дочь Нэнси активно участвовала в работе фонда. В 1970-е годы, когда стало очевидно, что для прогресса в работе необходимо лучше изучить генетику заболевания, она полностью взяла работу фонда в свои руки.
В Венесуэле есть озеро Маракайбо, на берегах которого царит жестокая нищета и к тому же гораздо чаще, чем обычно, встречается болезнь Хантингтона. В 1979 году Нэнси Векслер приступила к сбору образцов ДНК у крестьян и стала записывать родословные местных жителей, чтобы построить генеалогию всех местных жителей, пострадавших от этой болезни. Понимая, что в будущем недуг может поразить и ее, Нэнси Векслер заботилась о людях, живших в деревянных хижинах на сваях прямо над водой. При ходьбе эти люди раскачивались, как пьяные, точно как ее мать. Местные жители называли Нэнси Векслер «Катира» за длинные светлые волосы. Америко Негретте, ее венесуэльский коллега, впервые написавший о случаях болезни Хантингтона на берегах озера Маракайбо, рассказывает, что она превратила местных жителей в свою огромную семью, всякий раз приветствуя их «без наигранности и позерства, при встрече с венесуэльцами ее глаза просто лучились нежностью».
Цель экспедиций Нэнси Векслер заключалась в том, чтобы в конце концов найти ген, отвечающий за болезнь Хантингтона. Однако как генеалогия жителей Маракайбо могла помочь выявить виновника? Векслер и другие специалисты по генетическим болезням знали, что с людьми придется работать так же, как Морган и его ученики более века назад работали с дрозофилами. В эпоху секвенирования ДНК Векслер с коллегами могли отслеживать генетические маркеры в семейной родословной, то есть через несколько генетических скрещиваний, просто проанализировав ДНК в нескольких поколениях. Успехи в изучении патогенеза этой болезни начались за год до того, как Векслер приступила к своим генеалогическим исследованиям. Как и во многих других случаях научных прорывов, здесь не обошлось без интуитивной прозорливости.
В процессе работы над этой проблемой сформировался ежегодный ритуал: небольшая компания аспирантов из Университета штата Юта вместе с научными руководителями отправлялась на горный курорт Альта в горах Уосач-Маунтин на интенсивный семинар по своим профильным исследованиям (и, конечно, для того чтобы покататься на лыжах). Обычно к ним присоединялись двое-трое маститых ученых из других университетов, чтобы конструктивно покритиковать данные, предоставляемые каждым из волнующихся аспирантов. В 1978 году среди таких «тузов науки» были Дэвид Ботстейн из Массачусетского технологического института и Рон Дэвис из Стэнфорда.
Известные ученые, прибывшие на горный курорт, были весьма неординарными личностями. Дэвид Ботстейн был «склонен говорить и думать исключительно быстро, причем зачастую делал то и другое одновременно». Рон Дэвис – спокойный и сдержанный. В тот апрель в Юте Ботстейн и Дэвис, несмотря на полное несходство их темпераментов, обоюдно пришли к общему заключению. Они слушали доклад одного из аспирантов, ученика Марка Школьника – молодой человек с сожалением рассказывал о скудости генетических маркеров при бесплодном выискивании гена той или иной болезни. В этот момент Ботстейн и Дэвис обменялись взглядами и внезапно одновременно осознали истину. Хотя они специализировались на изучении дрожжей, оба одновременно догадались, как картировать человеческие гены! Новые ультрасовременные методы работы с рекомбинантной ДНК позволяли применить при изучении человека точно такой же генетический анализ, что уже использовался при картировании генов у других видов, но именно Ботстейн и Дэвис первыми реализовали на практике возможности этого метода в изучении генетики человека.
Методологическая находка Ботстейна и Дэвиса, ныне именуемая как «анализ сцепления», позволяет определить положение неизвестного генаотносительно известных генов-ориентиров. Принцип прост. Например, было бы непросто найти на карте США незнакомый для вас Спрингфилд, штат Масачусетс, если бы вы больше ничего не знали об этом городе, кроме названия. Если же я вам подскажу, что город Спрингфилд находится на половине пути между Нью-Йорком и Бостоном (двумя городами-ориентирами, обозначенными на карте), задача значительно упростится. Анализ сцепления позволяет проделать с генами то же самое: он выявляет связи между известными генетическими маркерами и неизвестными генами. Этот метод успешно зарекомендовал себя при работе с плодовыми мушками, использование его у человека представляло определенные трудности, поскольку у человека было известно очень мало генетических маркеров. Но Ботстейн и Дэвис осознали, что проблему возможно решить, используя достижения молекулярной биологии.
Маркерами в ДНК, на которые они обратили внимание, были те самые полиморфизмы длин рестрикционных фрагментов. ПДРФ, Restriction fragment length polymorphism, RFLP – способ исследования геномной ДНК путем разрезания ДНК с помощью эндонуклеаз рестрикции и дальнейшего анализа размеров образующихся фрагментов (рестриктов) путем гель-электрофореза (электрофореза ДНК). Напомню, что большинство ферментов-рестриктаз режут ДНК только в том месте, где встречают конкретную палиндромную последовательность. Если генетическая «буква» в этом месте изменится, то фермент больше не сможет разрезать ДНК в прежнем месте. Чаще всего эти повторы, которых в нашем геноме миллионы, встречаются в мусорной ДНК и потому в практическом отношении не имеют функционального эффекта.
После семинара в Альте Ботстейн, Дэвис и Школьник вместе с Рэем Уайтом, тогда работавшим в Университете штата Массачусетс, стали исследовать феномен полиморфизма длин рестрикционных фрагментов. В 1980 году на основе материала, полученного в процессе их сотрудничества, была написана эпохальная статья, открывшая новую страницу в молекулярной генетике человека. В статье был предложен четкий план того, как при помощи ПДРФ-маркеров можно построить карту маячков-маркеров в каждой человеческой хромосоме. Дэвид Ботстейн с коллегами вычислили, что 150 ПДРФ, равномерно распределенных по всему геному, вполне хватит, чтобы выявить мутантные гены, вызывающие болезнь. Собирая образцы ДНК в семьях, состоящих из нескольких поколений, в которых болезнь поражает предков и потомков, можно поочередно отследить закономерности наследования ПДРФ, отыскивая именно те гены, из-за которых болезнь сохраняется в семьях и которые показывают расположение мутантного гена.
В 1983 году Хелен Донис-Келлер (в ту пору жена Дэвида Ботстейна) основала Отдел генетики человека в бостонской компании Collaborative Research, Inc. Она стремилась построить карту сцепления ПДРФ в масштабах всего человеческого генома. Через четыре года после описываемых событий она опубликовала результаты своей работы в статье, метко названной «Карта генетических сцеплений в геноме человека». На этой карте было 403 маркера (гораздо больше, чем по исходной оценке Дэвида Ботстейна), охватывавших добрых 95 % генома. Та первая карта не была совершенной; например, некоторые хромосомы имели четко установленное место, расположение других подвергалось сомнению. Тем не менее возможность составления такой карты показала, что картирование всего генома осуществимо, что было важным достижением тех лет.
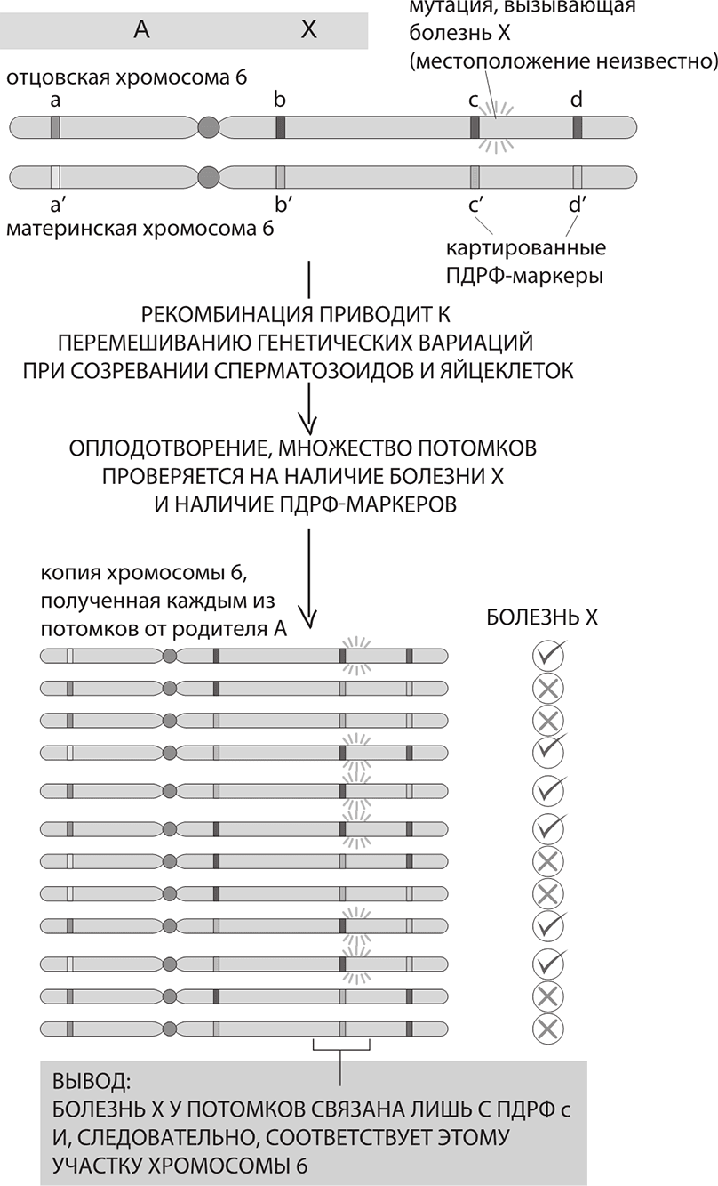
Генетическое картирование гена, отвечающего за болезнь. Для удобства показаны всего два поколения и лишь несколько индивидов. Чтобы анализ обладал статистической мощностью, в нем необходимо учесть большое число родственных индивидов
По мере того как работа по созданию подробной генетической карты набрала обороты, Дэвид Хаусман (David Houseman) из Массачусетского технологического института был готов начать работу, которую Ботстейн на данном этапе считал невыполнимым делом. Дэвид Хаусман поставил себе цель: локализовать ген болезни Хантингтона. Решить эту амбициозную задачу он поручил Джиму Гузелле, только что получившему звание PhD в лаборатории Хаусмана. Поначалу Ботстейн был настроен пессимистично, это было связано с отсутствием маркеров, необходимых для работы: ПДРФ хорошо выглядели только на бумаге, на тот момент работа по их сбору и анализу только началась. К 1982 году у Джима Гузелле было всего 12 ДНК-маркеров. Нэнси Векслер тем временем вернулась на Маракайбо и пыталась уточнить ранее собранную генеалогию: выяснить, кто и с кем вступал в брак, состояние здоровья их детей, кто кому приходился кузеном. Всего в одном семейном генеалогическом дереве, которое удалось выстроить Векслер, насчитывалось 17 тысяч имен! Помню одно из собраний, состоявшееся в Колд-Спринг-Харборе в октябре 1982 года, когда Джим Гузелла представил свои самые новые данные. Ни один из первых пяти маркеров не давал даже намеков на находку сцепления между генами, и меня не покидала мысль, что вся эта работа напоминает поиски иголки в стоге сена, а Джим Гузелла пока вытащил из стога всего несколько соломинок. Когда он завершил свою речь словами: «Обнаружение гена болезни Хантингтона – всего лишь дело времени», я про себя добавил: «Безусловно, только очень долгого времени».
Но удача любит смелых. К изумлению Гузеллы и всех прочих, двенадцатый маркер, обозначенный G8, оказался связан с болезнью Хантингтона. Впервые ген человеческой болезни удалось найти в хромосоме, не опираясь на данные о сцеплении с полом и не обладая никакими исходными знаниями о биохимической основе расстройства. Внезапно открылась новая научная перспектива: нам показалось, что в конце концов удастся проанализировать все те генетические дефекты, которые были бичом рода человеческого с начала времен его существования. Оказалось, что полиморфизмы длин рестрикционных фрагментов – очень действенный диагностический инструмент. После обнаружения гена болезни Хантингтона на конце короткого плеча 4-й хромосомы наши мощные методы клонирования генов, определенно, позволят выделить сам этот ген – это лишь дело времени, причем весьма недалекого.
Болезнь Хантингтона творит свое черное дело уже со взрослыми людьми. Мы знаем, что очень часто генетические болезни поражают детей, и они тем более ужасны, поскольку их жертвы практически не имеют шансов порадоваться этой жизни. Часто после постановки диагноза можно с суровой определенностью описать всю дальнейшую жизнь ребенка. Именно такая ситуация складывается с миодистрофией Дюшенна – прогрессирующим заболеванием, которое истощает мышцы, вызывает затруднения при движениях с детского возраста, которые прогрессируют с течением времени. Миодистрофия Дюшенна сцеплена с полом: вызывающая это заболевание мутация локализуется в X-хромосоме. Женщин, имеющих хромосому с такой мутацией, обычно спасает от болезни вторая, нормальная копия этого гена в другой X-хромосоме. Однако если хромосома с мутантным геном передается сыну, то мальчик заболеет миодистрофией Дюшенна, так как не имеет второй X-хромосомы с нормальной копией гена. Когда ребенку будет около пяти лет, родители заметят, что ему сложно встать с пола или подняться по лестнице. Примерно к десяти годам он окажется в инвалидной коляске. Вероятнее всего, больной умрет в подростковом возрасте либо проживет чуть больше двадцати лет.
В конце 1970-х годов цитогенетики (ученые, изучающие хромосомы под микроскопом) обнаружили аномалию в коротком плече X-хромосомы, встречающейся у тех считаных девочек, которые подвержены миодистрофии Дюшенна. Речь идет об участке Xp21. Анализ сцепления, выполненный Бобом Уильямсоном из Медицинской школы при больнице Святой Марии в Лондоне, а также его коллегой Кей Дэвис (в замужестве Дейм), подтвердил, что все дело в участке Xp21. Тем временем генетики-клиницисты воспользовались полиморфизмами длин рестрикционных фрагментов в качестве диагностического инструмента, позволяющего определить, кто из членов той или иной семьи (не исключая этапа внутриутробного развития) несет такую мутацию. Например, если у женщины есть мутация, провоцирующая миодистрофию Дюшенна, то ее сын с вероятностью 50 % заболеет этим недугом. Впервые в истории с помощью метода ПДРФ врачам удалось внутриутробно определить риск рождения ребенка с генетическим заболеванием.
Такой метод диагностики впервые представили в 1978 году сотрудники Калифорнийского университета в Сан-Франциско Юэт Кан и его коллеги. Они использовали сцепленные ПДРФ для диагностики плода с β-талассемией. ДНК для анализа забиралась либо из амниотической жидкости, в которой присутствуют клетки плода, либо из плаценты. Такая процедура называется «биопсия хориона». Правда, точность этого метода не составляла 100 %, поскольку, если вы помните из первой главы, при возникновении яйцеклеток хромосомы подвергаются рекомбинации. Если такая перестановка генетического материала происходит между ПДРФ и интересующим нас геном, то результат биопсии получится неверным, отсюда и погрешность. Она составляет при раннем обнаружении миодистрофии Дюшенна около 5 % и является неизбежным следствием рекомбинации. Но результаты пренатальной диагностики целиком и полностью зависели от выявления конкретного гена, а не от маячковых маркеров.
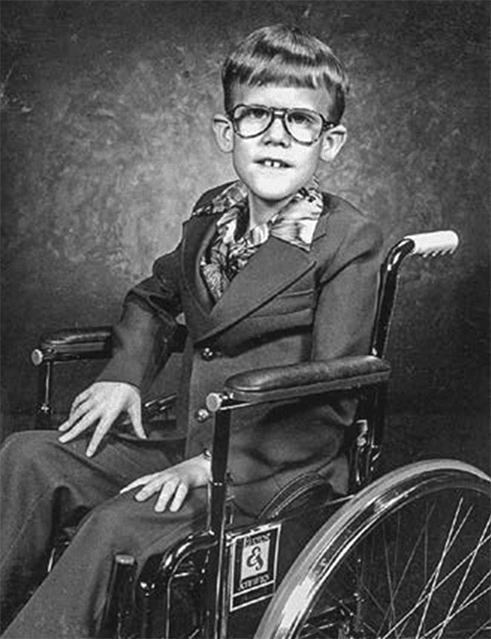
Брюс Брайер, чья частичная делеция X-хромосомы позволила идентифицировать ген миодистрофии Дюшенна. Брюс погиб в автокатастрофе в возрасте семнадцати лет, но прожил относительно полноценную жизнь и даже был отличным органистом.
Ген миодистрофии Дюшенна удалось идентифицировать благодаря мальчику по имени Брюс Брайер, у которого в коротком плече X-хромосомы не хватало очень большого фрагмента. Этот участок был таким большим, что Брюс страдал не только миодистрофией Дюшенна, но и еще тремя другими генетическими заболеваниями. В 1985 году Лу Кункель из Гарвардской медицинской школы рассудил, что мог бы воспользоваться ДНК Брюса, чтобы выделить нормальный ген из ДНК здорового мальчика. Кункель осознал, что вся ДНК Брюса должна быть и у здорового мальчика, а те фрагменты, которые есть у здорового мальчика, но отсутствуют у Брюса, и несут разгадку. Воспользовавшись рекомбинантной технологией, Лу Кункель извлек из нормальной ДНК ту часть, которая была у Брюса, и стал изучать отличающиеся фрагменты, по его мнению, именно в них должен был находиться ген миодистрофии Дюшенна.
Полностью тайна заболевания была разгадана Тони Монако, аспирантом Лу Кункеля. Оказалось, что последовательность pERT87 отсутствует у пятерых мальчиков, страдающих миодистрофией Дюшенна; она располагалась очень близко к искомому гену, иногда даже входила в его состав. Теперь гену уже можно было дать подходящее название: дистрофин. Несколько лет этот ген считался самым крупным в геноме человека, поскольку в нем очень много больших интронов, но затем дистрофин уступил первенство гену, кодирующему другой мышечный белок и не менее удачно названному «титин».
Эти новые знания были немедленно использованы для пренатальной диагностики миодистрофии Дюшенна. Однако, несмотря на то что функция дистрофина была исследована на протяжении десятилетий, мы по-прежнему не в силах эффективно лечить или облегчать течение миодистрофии Дюшенна. Именно это удручает в текущем положении дел: генетика позволяет идентифицировать и понять болезнь, но пока в большинстве случаев не позволяет исправить генетические ошибки. Подобные истории связаны и с болезнью Хантингтона, и с муковисцидозом, и с другими наиболее серьезными генетическими расстройствами. По сравнению с разработкой препарата, позволяющего блокировать известную мишень, лечение генетического заболевания – вызов совершенно иного плана: здесь требуется добавить недостающие гены или заменить нефункциональные. В случае с миодистрофией Дюшенна речь идет как раз о том, как доставлять достаточно крупный белок к мышцам.
Среди некоторых наиболее перспективных методов борьбы с миодистрофией Дюшенна рассматривается возврат к методу генетической терапии, который ранее считался совершенно безнадежным, об этом мы поговорим далее в главе. Исследователи пробуют организовать доставку дистрофина в мышечные волокна при помощи вирусов – речь идет либо о полноценном гене, либо о его уменьшенной версии. Другой метод больше подходит тем немногим пациентам с миодистрофией Дюшенна, которые страдают от иной мутации: она преждевременно блокирует трансляцию транспортной РНК дистрофина. Биотехнологические компании, например PTC Therapeutics, разрабатывают препараты, позволяющие рибосоме миновать ложные стоп-сигналы для получения полноценной коммуникации, на основе которой выстраивается полноценный белок. В 2016 году компания Sarepta Therapeutics получила одобрение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) на препарат этеплирсен, увы, это лекарство предназначено для лечения немногих пациентов, а его клиническая эффективность остается до конца невыясненной. Наконец, Кей Дэвис и ее коллеги из Оксфордского университета разработали метод замены недостающего дистрофина, искусственно усиливая экспрессию родственного гена, который называется утрофин. Этот метод хорошо работает на мышиных моделях с миодистрофией Дюшенна, пока ведутся клинические испытания.
Наиболее интенсивные поиски генов наследственных болезней пришлись на 1980-е годы, когда врачи пытались побороть одно из самых распространенных генетических заболеваний – муковисцидоз. Охота за геном муковисцидоза вышла особенно примечательной по двум причинам: впервые коммерческая компания участвовала в картировании гена человеческой болезни, и впервые развернулось жестокое соперничество среди ученых, причастных к этому направлению исследований.
При муковисцидозе в легких у больного скапливается густая слизь, затрудняющая дыхание. Клетки, выстилающие легочные трубки, не могут элиминировать слизь, в которой размножаются бактерии, поэтому человек страдает от инфекций верхних и нижних дыхательных путей. До открытия антибиотиков ожидаемая продолжительность жизни при муковисцидозе составляла не более десяти лет; сегодня выживаемость значительно увеличилась, многие пациенты разменивают свой четвертый и даже пятый десяток. В Северной Европе муковисцидоз встречается с частотой примерно один случай на 2500 человек. Эта болезнь наследуется по рецессивному принципу: человек заболевает, лишь если у него сразу две мутантные копии гена. Мутантный ген муковисцидоза встречается примерно у каждого 25-го жителя Северной Европы, то есть на удивление часто. Существует гипотеза, что его носители обладают селективным «преимуществом гетерозигот», например как защита от малярии, которую дает серповидноклеточная анемия. Вероятно, муковисцидоз уберегал носителя от инфекционных заболеваний, свирепствовавших в Европе в XVII–XVIII веках, например от туберкулеза и холеры.
Лап-Чи Цуи родился в Шанхае, вырос и выучился в Гонконге, а в США приехал в 1974 году, уже будучи аспирантом. Цуи освоил молекулярную генетику, изучая вирусы, а затем в 1981 году перебрался в Торонто, где стал работать над проблемой муковисцидоза, кропотливо выискивая любые ПДРФ в семьях, страдающих этой болезнью. Но не только Цуи искал ген муковисцидоза: в то же самое время поисками занимались Боб Уильямсон в Лондоне и Рэй Уайт в Юте, в их распоряжении была подробная генеалогическая информация, собранная церковью мормонов. Метрики мормонов, именуемые «Анналы предков», позволяют их современным потомкам отдать должное почившим предкам, прожившим жизнь вне церкви или умершим до ее основания (церковь мормонов существует с 1830 года). Подробная запись данных у мормонов преследовала вполне определенную цель – воссоединить семьи в вечности. Для нас это тот самый случай, когда нужды генетики и религии так замечательно совпали.
Лап-Чи Цуи добился первых успехов в 1985 году, когда нашел связь между патентованным набором ПДРФ, открытым для использования с целью анализа генов муковисцидоза, и непосредственно самим геном муковисцидоза. Уильямсон и Уайт буквально дышали ему в затылок. Их статьи были одновременно опубликованы в журнале Nature, и в этих работах сообщалось, что ген муковисцидоза локализован в длинном плече седьмой хромосомы, а ближайший маркер отстоит от него примерно на миллион оснований.
Следующий этап обещал быть еще тяжелее, если вы помните, поиски происходили за пять лет до запуска проекта «Геном человека». Миллион оснований по-прежнему оставался огромным промежутком для клонирования «генетиков-навигаторов». Поэтому Лап-Чи Цуи объединился с Френсисом Коллинзом, экспертом по молекулярной генетике, который в ту пору работал в Мичиганском университете, а впоследствии успешно сменил меня на посту директора проекта «Геном человека».

Лап-Чи Цуи, искатель генов
Френсис Коллинз разработал методы «прыжков» между двумя известными полиморфизмами длин рестрикционных фрагментов. В 1989 году, после двух лет работы, Цуи и Коллинз смогли идентифицировать кандидатный ген, кодирующий мембранный белок, известный своей ролью в функционировании потовых желез у человека; известно, что у больных муковисцидозом эти железы не работают. Секвенирование гена и поиск мутаций в обеих его копиях у пациентов с муковисцидозом завершились успехом: оказалось, что у большинства больных отсутствует участок ДНК длиной в три основания, а в достаточно большом по размерам белке муковисцидоза не хватает всего одной аминокислоты. Такого дефицита аминокислоты оказалось достаточно, чтобы полностью нарушить естественную укладку белка и доставку его к клеточной мембране. На эту единственную мутацию приходится 70 % всех случаев муковисцидоза. Кроме описанного дефекта зарегистрировано еще более тысячи связанных с ним мутаций. Такое изобилие патологических вариантов значительно усложнило гено-диагностику муковисцидоза.
В то время как генетическое сообщество праздновало удачное картирование и выделение гена муковисцидоза, Нэнси Векслер, Дэвид Хаусман, Джим Гузелла и их коллеги все занимались поисками гена болезни Хантингтона. Ушло десять лет работы, в которой участвовало 150 ученых со всего мира, пока «зловредный» ген наконец не был засечен: консорциумученых выделил его на критически важном участке 4-й хромосомы в позиции 4p16.3, который назвали IT15 («интересный транскрипт 15»). Генетический дефект, приводящий к хорее Хантингтона, обусловлен экспансией нестабильного тринуклеотидного кодона CAG, кодирущего глутамин, который транслируется в протеине как полиглутаминовый повтор. У здорового индивида встречается меньше 35 последовательных CAG, а у пациентов с болезнью Хантингтона таких фрагментов множество. CAG – это генетический код аминокислоты глутамина. У страдающих болезнью Хантингтона структура белка хантингтина может быть самой разной, так как из-за полиморфизма гена в белке содержится разное количество остатков глутамина. Молекулярная масса хантингтина в основном зависит именно от количества остатков глутамина. Вероятно, такое различие влияет на работу белка в клетках мозга; его молекулы могут образовывать внутри клетки «липкие белковые комочки», и из-за этого клетка погибает. Такое любопытное увеличение последовательностей повторяющихся тринуклеотидов лежит в основе патогенеза многих других неврологических заболеваний, в том числе при синдроме фрагильной X-хромосомы и при спиноцеребеллярной атаксии (spinocerebellar ataxia, SCA). Однако мы до сих пор не вполне понимаем, почему клетки мозга становятся уязвимы для подобных причудливых мутаций.
Несмотря на то что на поиск генов, отвечающих за генетические расстройства – болезнь Хантингтона, миодистрофию Дюшенна и муковисцидоз, – уходит немало времени, по генетическим меркам это «простые» болезни. Они возникают из-за мутации единственного гена и практически не зависят от условий окружающей среды. Таких расстройств существует огромное число, только в современной генетической базе данных перечислено более тысячи, хотя общепопуляционно их число невелико, и каждое из них встречается лишь в отдельных семьях.
Гораздо более распространены «сложные», или полигенные, расстройства, к которым относится множество самых известных недугов: астма, шизофрения, депрессия, врожденные пороки сердца, гипертензия, диабет и рак. Они возникают из-за взаимодействия множества генов, каждый из которых в отдельности оказывает лишь незначительный эффект. Причем для полигенных расстройств обычно характерна еще одна проблема: такие группы взаимодействующих генов могут провоцировать предрасположенность к определенному заболеванию, но разовьется оно или нет, зависит от условий окружающей среды. Допустим, у вас есть набор генетических вариантов, предполагающий склонность к алкоголизму. Однако ваша судьба в этом направлении, а именно ответ на вопрос, станете ли вы алкоголиком, зависит от того, как вы отреагируете на источник возможной болезни, то есть на алкоголь. То же касается и астмы: «хорошим» летом, когда в воздухе мало пыльцы и спор, никаких симптомов астмы у вас может и не быть, несмотря на генетическую предрасположенность к этому заболеванию.
Сложные взаимодействия генов и окружающей среды наиболее очевидны в онкологии. Как будет рассказано в главе 14, рак по сути своей – генетическое расстройство, возникающее из-за мутаций в нескольких генах и в итоге приводящее к возникновению злокачественных клеток. Есть два варианта возникновения раковых мутаций. Некоторые передаются по наследству. Всем известна фраза «это у них семейное», и если некоторые социальные признаки, характеризуемые таким образом, например католицизм, не всегда передаются по наследству, некоторые виды рака неизбежно наследуются. Множество раковых мутаций возникает и в ходе нормальной жизнедеятельности. ДНК может быть повреждена из-за ошибок в работе ферментов, отвечающих за репликацию или починку генетической молекулы, либо вследствие побочных эффектов при нормальных химических реакциях в клетке. Многие виды рака возникают из-за наших же глупых пристрастий к канцерогенам: например, нам нравится жариться под ультрафиолетом или выкурить сигарету. Смысл в том, что ДНК легко повреждается, но именно мы в силах свести эти поломки к минимуму, осознанно организуя свою личную и социальную жизнь.
В 1974 году Мэри-Клэр Кинг (прославившаяся изучением родства между человеком и шимпанзе и работой с «Лас Абуэлас») перебралась в Калифорнийский университет в городе Беркли, где целиком посвятила себя поискам гипотетического гена, вызывающего рак груди. На тот момент до разработки метода, использующего полиморфизм длин рестрикционных фрагментов, оставалось еще шесть лет, но Кинг взялась собирать информацию о семьях, у членов которых в раннем возрасте развивался рак груди (и яичников). Она рассудила, что эти заболевания вполне могут иметь наследственную природу. Скептики были убеждены, что рак груди слишком сильно зависит от факторов окружающей среды и генетический анализ в данном случае бесполезен. Непоколебимая Мэри-Клэр Кинг продолжала уточнять свой набор данных и к 1988 году, проанализировав более 1500 семей, собрала доказательства в пользу того, что ген предрасположенности к раку груди действительно существует.
Завершив анализ сцепления более сотни маркеров ДНК, Кинг поразила все медицинское сообщество, когда в 1990 году объявила, что нашла в 17-й хромосоме участок полиморфизма длин рестрикционных фрагментов, ассоциированный с раком груди в подгруппе, состоящей из 23 семей ее выборки. На эти 23 семьи суммарно пришлось 146 случаев рака груди. Прислушавшись к предположению одного из своих аспирантов, Мэри-Клэр Кинг совершила открытие: в тех семьях, где рак груди начинал развиваться в сравнительно юном возрасте, вероятность наследования гена предрасположенности к этому виду рака была выше. Таким образом, выяснено, что ген расположен в хромосоме на участке 17q21 и при его мутации риск заболеть раком груди у женщины сильно возрастает. После публикации статьи Кинг в журнале Science в 1990 году началась гонка – кто первым выделит сам этот ген, названный BRCA1 (ген кодирует рак груди первого типа), а также развернулись не завершившиеся до сих пор споры о коммерческом использовании генов.
Выделение гена BRCA1 неизбежно обещало быть событием планетарного масштаба. Мэри-Клэр Кинг стала работать вместе с Френсисом Коллинзом, незадолго до этого идентифицировавшим ген муковисцидоза. В сентябре 1992 года представительница одной большой семьи со множеством случаев рака груди, назову ее Анна, рассказала Барбаре Уэбер, сотруднице Коллинза, что ей назначена профилактическая мастэктомия, хотя у нее не было никаких признаков рака. Не в силах больше выносить неопределенность, Анна решилась на такой радикальный превентивный шаг. Однако Барбара Уэбер сделала ей анализ сцепления ДНК и пришла к выводу, что Анна не унаследовала патологического гена BRCA1: она рисковала заболеть раком груди не больше, чем любая другая женщина, не имеющая такого семейного анамнеза. Но этот вывод был сделан в контексте ведущегося исследовательского проекта, причем было заранее оговорено, что такие предварительные данные не будут использованы для клинической диагностики.
Однако Уэбер и Коллинз решили, что просьба Анны перевешивает такую регламентацию. Они сообщили женщине, что та почти не рискует, и она с огромным облегчением отказалась от операции. Неудивительно, что исследователи чувствовали себя обязанными оказать такую же услугу и другим членам ее семьи, если те их об этом попросят. Одна из женщин этой семьи, которая, как оказалось, тоже практически не рисковала, уже перенесла профилактическую двустороннюю мастэктомию пятью годами ранее. Она отнеслась к долгожданному диагнозу философски: рассудила, что благодаря операции смогла пять лет прожить спокойно. Сегодня доказано, что профилактическая мастэктомия действительно снижает смертность от рака среди женщин, входящих в группу риска; аналогично при удалении яичников в возрасте до сорока лет снижается вероятность как рака яичников, так и рака груди. Генетический анализ может помочь женщине принимать решения, которые для нее в буквальном смысле являются вопросом жизни и смерти.
Кинг и Коллинз столкнулись с жесткой конкуренцией в погоне за геном BRCA1. Марк Школьник из штата Юта, генетик, участвовавший в прорывных исследованиях сцеплений, совместно с Уолли Гилбертом основал компанию Myriad Genetics (Уолли Гилберт еще не растерял предпринимательского духа даже после непростых времен, пережитых в компании Biogen). Бизнес-план Myriad был таков: воспользоваться массивом данных о родословной мормонов и на основе этой информации картировать и изолировать ген BRCA1. Myriad Genetics одержала победу в четырехлетней гонке, опередив конкурентов на считаные недели и опубликовав в журнале Science статью об эпохальном открытии гена рака груди BRCA1. Во главе команды поисковиков из Myriad стоял молодой биолог-молекулярщик Александер (Саша) Кам, внук великого Лайнуса Полинга. Это был сокрушительный удар для Мэри-Клэр Кинг, но она и не думала отступать, продолжая дополнять длинный список мутаций BRCA1, зафиксированных в тех семьях, которые она изучала. В 1997 году была одобрена патентная заявка Myriad Genetics на этот ген, и компания воспользовалась этим, чтобы стать монополистом в области анализов на BRCA1. Не менее интенсивная гонка развернулась при поисках второго гена рака груди BRCA2, локализованного в 13-й хромосоме. На этот раз группа ученых из Института онкологических исследований в Великобритании заявила, что обогнала Myriad Genetics, но патент на данную последовательность успели подать обе группы.
Уже был понятен коммерческий потенциал этих генов. Риск возникновения рака груди к семидесятилетнему возрасту у носительницы гена BRCA1 или BRCA2 из-за накопления мутаций в этих генах достигал 80 %. Более того, те же самые мутации в 45 % случаев также повышают риск возникновения рака яичников. Женщины из семей, в которых наследуются такие мутации, должны как можно раньше узнавать, есть ли у них дефектный вариант первого или второго гена. Широко известно, на какие действия пошла актриса Анджелина Джоли: в таких случаях приходится делать мучительно тяжелый, но потенциально спасительный выбор. Выборочная двусторонняя мастэктомия у женщин, подверженных высокому риску возникновения рака груди, позволяет снизить число случаев такого рака на 90 %. В то же время генетический скрининг помогает выявить в таких семьях индивидов с нормальными генами; и женщина, у которой нет мутации, сможет успокоиться, осознав, что никакого повышенного риска для нее нет.
За двадцать лет Myriad Genetics помогла тысячам женщин принять информированные решения о здоровье, возможно, спасшие многим жизнь. Тем не менее компанию из Солт-Лейк-Сити часто приводят в качестве примера порочной практики, возникающей, когда коммерция подмешивается к науке. Ведь как ни рассуждай, компания Myriad десять с лишним лет наживалась на своей глобальной монополии на анализ гена BRCA1, заявляя, что вправе зарабатывать деньги таким образом, чтобы вернуть миллионы долларов, потраченные на поиск генов и разработку анализа. Вопрос надоставить по-другому. Сколько денег вправе таким образом заработать компания? Ее тестирование на наличие наследственного рака молочной железы и яичников (BRAC Analysis) стоит более трех тысяч долларов, при том что Myriad Genetics вдобавок к этому держала в строгой тайне обширную базу данных по мутациям BRCA, собранную ими за двадцать лет.
После нескольких бурных разбирательств между компаниями с пациентами в мае 2009 года Американский союз защиты гражданских свобод (ACLU) подал от имени этих нескольких пациентов иск против компании Myriad Genetics и Ведомства по патентам и товарным знакам США. Как уже было рассказано мной в главе 8, в 2013 году Верховный суд единогласно постановил, что успех Myriad, позволивший компании почти двадцатью годами ранее выделить ген BRCA1 и разработать анализ для его диагностики, нельзя считать изобретением. Такой вердикт сразу же развязал руки другим диагностическим компаниям, в том числе Ambry Genetics, Gene by Gene и Pathway Genomics, которые стали оспаривать монополию Myriad и запустили производство собственных бюджетных исследований, проверявших сразу два гена: BRCA1/2. В настоящее время все (или практически все) юридические вопросы между сторонами улажены.
В 1990-е годы, одновременно с разработкой проекта «Геном человека» анализ сцепления генов помог выявить ряд других раковых генов, в том числе вызывающих нейрофиброматоз (болезнь, которой страдал Джозеф Кэри Меррик, «человек-слон»), рак прямой кишки и рак простаты. Также были картированы гены, вызывающие редкие наследственные формы болезни Альцгеймера и болезни Паркинсона. Тем не менее при всей внешней эффективности поиск по принципу «ген за геном» получался медленным и трудоемким, каждое исследование зависело от того, удастся ли найти подходящие семьи для анализа. Существует и другая стратегия: искать небольшие изолированные популяции, где та или иная болезнь встречается очень часто. В природе сложно найти более крошечную и изолированную популяцию, чем население острова Тристан-да-Кунья.
Тристан-да-Кунья – это вулканический остров, его крутые и негостеприимные берега вздымаются посреди моря. Он представляет собой просто клочок суши площадью всего 98 квадратных километров посреди Южной Атлантики и считается одним из самых отдаленных мест на планете. Первое постоянное поселение на острове состояло из служащих британского гарнизона, который высадился на него в 1816 году, чтобы помешать французским войскам устроить там базу для вызволения императора Наполеона из ссылки на острове Святой Елены, расположенном на расстоянии 2,5 тысячи километров к северу. В дальнейшем население острова росло эпизодически: в какой-то части острова обосновались немногочисленные поселенцы, где-то – выжившие после очередного кораблекрушения. По неофициальной переписи 1993 года, численность населения острова составляла всего 301 человека. Именно в тот год группа сотрудников Торонтского университета отправилась на остров, чтобы изучить отдаленные результаты медицинских исследований, в которых островитяне участвовали еще в 1961 году, когда из-за проснувшегося на острове вулкана все население острова временно эвакуировали в Англию. Самым удивительным оказалось то, что в анамнезе более чем у половины беженцев была астма.

Пожалуй, самая отдаленная обитаемая территория на планете: вид на остров Тристан-да-Кунья с близлежащего необитаемого острова
Современное исследование ученых из Торонто, включавшее 282 островитянина, также показало, что у 161 человека (57 %) наблюдаются симптомы астмы. Канадцы подняли генеалогию всех местных семей – потомков первых 15 поселенцев. По всей видимости, астму занесли на остров две женщины, обосновавшиеся здесь в 1827 году, после чего население значительно возросло, но фактически представляло собой одну расширенную семью. В более крупной и смешанной популяции астма, вероятно, могла бы быть вызвана различными наборами аллелей, но на острове такого события произойти не могло в силу изолированности. Именно из-за разнородности тяжело выделить генетические первопричины сложных заболеваний. Остров дал фантастические возможности для проведения исследований.
Далее команда медиков из Торонто стала работать совместно с компанией Sequana из Сан-Диего, основанной специально для поиска генов, вызывающих патологические процессы. Впоследствии представители Sequana заявляли, что обнаружили в 11-й хромосоме два гена, провоцирующих предрасположенность к астме. Далее было проведено исследование, в ходе которого были изучены примерно 2 миллиона фрагментов у тысяч астматиков; исследование позволило предположить, что наиболее распространенные генетические факторы риска связаны с 17-й хромосомой, а конкретно – с иммунными факторами. Тем временем канадские активисты заявили, что компания совершила «акт биопиратства», «нарушив основополагающие права тех людей, у которых брали образцы ДНК», то есть принцип добровольного информированного согласия. Пока до социального извержения было далеко, но первый толчок в обществе уже произошел.
Буря, разразившаяся по поводу «биопиратства» Sequana, не шла ни в какое сравнение с ураганом, налетевшим на Кари Стефанссона (Kári Stefánsson) и его компанию deCODE Genetics несколько лет спустя. Кари Стефанссон решил, что для исследования тоже нужно найти отдаленный остров, но более густонаселенный, чем Тристан-да-Кунья, у обитателей которого можно было бы сразу определить несколько патологических генов. Так сложилось, что Кари Стефанссон родился именно на таком острове.
Родина Кари Стефанссона – Исландия, по площади примерно как штат Кентукки, но население всей страны составляет примерно 323 тысячи человек, что в 13 раз меньше, чем в этом штате. В IX–X веках остров заселили викинги, которые привезли с собой женщин, похищенных в Ирландии во время набегов. Поэтому Исландия была интересна для предприимчивого охотника за генами сразу по нескольким причинам. Во-первых, население Исландии исключительно однородно: практически все жители происходят от тех самых первых поселенцев, со времен викингов в страну мало кто иммигрировал. Во-вторых, имеются метрики всех родившихся в стране жителей с 1838 года, кроме этого реестра, есть и подробные генеалогические архивы, уходящие в прошлое на много поколений. Стефанссон утверждал, что может проследить собственную родословную на тысячу лет в прошлое, вплоть до легендарного исландского поэта и воина Эгилля Скаллагримссона, одного из героев исландских саг. В-третьих, в Исландии с 1914 года действует государственная служба здравоохранения, поэтому медицинские карты всей нации есть в наличии, они хранятся в строгом порядке, и их можно изучать.
Кари Стефанссон, невролог с гарвардским дипломом, интересовался сложными генетическими расстройствами, в частности рассеянным склерозом и болезнью Альцгеймера. Понимая, что его соотечественники – идеальная популяция для подобных исследований, он стал сооснователем компании deCODE и взялся за соотношение реестров медицинских (уже имеющихся) с реестрами генетическими. Стефанссон решил собрать максимально полную базу данных для поиска генов. На это у него имелось официальное разрешение от исландского парламента.
В 2000 году deCODE получила двенадцатилетнюю лицензию на создание и эксплуатацию Исландской медицинской базы данных. Генеалогические сведения были общедоступны, но в базу данных попадала информация из медицинских карт лишь тех островитян, которые на это согласились. В дальнейшем такие заверения жителей не спасли deCODE от широкого общественного осуждения относительно приватности генетических данных.
Большинство исландцев одобрили миссию компании, а также потенциальный положительный эффект для небольшой экономики своей страны. После завершения проекта «Геном человека» компания deCODE стала одной из самых успешных и плодовитых в плане открытий генетических организаций, открывшей аллели, ответственные за развитие десятков сложных, генетически ассоциированных нарушений: сердечных заболеваний, остеопороза, депрессии, шизофрении, инсульта и рака. Компания взяла на вооружение технологию ДНК микрочипов, позволившую генетикам отследить сотни тысяч однонуклеотидных полиморфизмов в рамках единого исследования ассоциаций, охватывающего весь геном. Момент истины для полногеномного поиска ассоциаций (GWAS) наступил в 2007 году, когда в журнале Nature была опубликована знаковая статья от фонда Wellcome Trust Сенгеровского института. В этой статье были опубликованы результаты исследования, проведенного с участием около 17 тысяч человек. Результатом этого исследования было выявление аллелей, вызывающих семь распространенных заболеваний, в том числе депрессию, болезнь сердца и болезнь Крона. На сегодняшний день уже существуют воспроизводимые опыты, доказывающие ассоциацию более 10 тысяч вариантов всевозможных патологических состояний с генетическими признаками: кардиологических расстройств, фибрилляции предсердий, многих видов рака, артрита, волчанки, целиакии, причем этот список далеко не полон.
Одновременно со стремительным падением стоимости полногеномного секвенирования и ее приближением к отметке в тысячу долларов росли амбиции ученых, планировавших крупномасштабные популяционные исследования. Речь шла уже не об идентификации отдельных генов, а об обеспечении проекта комплексного геномного здравоохранения. Примеры таких проектов ранее уже существовали: рассмотренный выше FarGen (Фарерские острова), Genomics England (финансируемая правительством Великобритании программа по секвенированию ДНК 100 тысяч пациентов), Precision Medicine Initiative (Инициатива в сфере точной медицины) в США. Первое поколение исследователей генома, включающее академические и коммерческие организации, зачастую сосредоточивалось на изучении одного гена или одного заболевания. Сегодня секвенирование ДНК и анализ геномов настолько усовершенствовались, что медицинские и фармацевтические компании могут вынашивать идеи крупномасштабных проектов по секвенированию геномов сотен тысяч человек в поискахмельчайших генетических мутаций, влияющих на наш разум, состояние организма и долголетие.

В 2015 году президент Обама официально объявил в Белом доме о старте проекта «Инициатива в сфере точной медицины»
Хотя сегодня мы значительно лучше представляем себе генетическую подоплеку распространенных болезней, чем до реализации проекта «Геном человека», пока мы выиграли лишь одну битву, но не войну в целом. С 2007 года были достоверно установлены ассоциации примерно с сотней генов и других участков генома, при которых у человека могут развиться воспалительные заболевания кишечника. Эта информация оказалась очень полезной, но встал другой вопрос: какие гены наиболее важны? Теперь исследователям требовалось проанализировать список наиболее актуальных и интересных «генов-подозреваемых», и практически каждый из этих генов может стать потенциальной терапевтической мишенью. Хуже того, почти половина ассоциаций, которые можно воспроизвести экспериментально, находятся вне генов, в плохо изученных «пустынных» регионах генома. В некоторых случаях такие генетические варианты нарушают работу регуляторных участков, влияя на работу генов, расположенных очень далеко. Более того, совокупность всех генов, которые ассоциированны с тяжелыми соматическими заболеваниями, например болезнью Крона или диабетом, составляет малую долю всех генетичеких изменений у конкретного человека. Эта досадная неопределенность часто именуется термином, напоминающим заглавие детективного романа: «скрытая наследуемость». Мне грустно об этом говорить, но генетические находки не так хорошо помогают нам понять биологию заболеваний, как нам хотелось. Еще один пример – шизофрения, проблема, крайне актуальная для моей семьи. Однако генетические исследования этой болезни весьма сумбурны. В 1988 году группа лондонских ученых объявила, что в ходе анализа сцеплений им удалось картировать доминантный ген шизофрении, расположенный в 5-й хромосоме. Результаты этого исследования были опубликованы в журнале Nature, равно как и результаты дальнейших исследований, безапелляционно развенчавших это заявление. Давайте перенесемся вперед на двадцать пять лет: представители нового поколения исследователей объединились, чтобы проанализировать миллионы последовательностей у тысяч пациентов в поисках таких аллелей, которые связаны с риском развития шизофрении. На начальном этапе исследований в нем участвовало шесть тысяч пациентов, затем 20 тысяч, но увеличение испытуемых не приблизило нас к пониманию проблемы. Расширенное исследование 110 тысяч больных и добровольцев из контрольной группы показало, что на подобные ассоциации указывает около сотни маркеров ДНК. Наиболее сильный маркер связан с геном четвертого фактора системы комплемента (C4), более известного своей ролью в реализации протективных свойств врожденного иммунитета, но, что весьма интересно, выполняющего совершенно иную функцию, приводя к мутациям, изменяющим нейронную сеть и влияющим на синаптическую пластичность, необходимое условие для обучения и памяти. По-видимому, чем активнее этот ген, тем выше риск развития шизофрении.
Вполне возможно, что это и есть ключевая подсказка о сути патогенетического пути развивития шизофрении, но позволит ли она фармацевтам подступиться к созданию лекарства от этой болезни? Я отчаянно надеюсь, что так и будет. Все, о чем я сейчас говорю, всего лишь одно из проявлений фундаментальной проблемы, которая касается исследования всевозможных генетических заболеваний: как редких, так и распространенных. Как только мы найдем все или почти все аллели, обусловливающие эти болезни, возникнет другой вопрос: что делать дальше?
Иногда генетические болезни можно излечить без помощи новейших препаратов или специальной терапии, а просто поняв базовую причину такого заболевания. Возьмем, к примеру, один из наиболее изученных врожденных дефектов метаболизма, тот самый, из-за которого на некоторых продуктах, особенно на бутылках с газировкой, мелким шрифтом предупреждают: содержит фенилаланин. Фенилаланин – это аминокислота (один из основных первокирпичиков, слагающих белки), которая не усваивается людьми с генетическим расстройством под названием фенилкетонурия (ФКУ).
Эта история началась в Норвегии в 1934 году. Молодая мама была полна решимости выяснить, что же не так с двумя ее детьми четырех и семи лет, которые, казалось бы, родились совершенно здоровыми. Старший был плохо приучен к горшку и едва выговаривал несколько слов, а уж о полноценных предложениях не шло и речи. Врач и биохимик Асбьерн Феллингвыявил у этих детей любопытную биохимическую аномалию: у них в моче было слишком много фенилаланина. Асбьерн Феллинг обнаружил еще тридцать четыре таких случая в двадцати двух семьях по всей Норвегии и понял, что столкнулся с генетическим заболеванием.
На сегодняшний день уже известно, что ФКУ обусловлена мутацией в гене фенилаланингидроксилазы – это фермент, преобразующий фенилаланин в другую аминокислоту, тирозин. Это редкое наследственное заболевание, имеющее аутосомно-рецессивный путь передачи, в Северной Америке встречается примерно 1 раз на 10 тысяч человек. У детей, страдающих этой болезнью, фенилаланин накапливается в крови, тормозит развитие мозга и приводит к тяжелому отставанию в умственном развитии. Предотвратить заболевание просто: дети с ФКУ вырастают нормальными, если с самого рождения живут на диете, бедной фенилаланином, то есть с минимальным содержанием белка и без искусственно подслащенных напитков. Принципиально важно как можно раньше, лучше после рождения выяснить, рискует ли ребенок заболеть ФКУ. Роберт Гатри разработал простой анализ крови, позволяющий определить уровень фенилаланина, и неустанно продвигал его, пока анализ не вошел в стандартную практику медицинского скрининга. С 1966 года в США у каждого новорожденного берут анализ крови из пятки и проверяют уровень фенилаланина. Тест Гатри, при котором не проверяется ни единой «буквы» ДНК, ежегодно позволяет выявить у миллионов младенцев редкие генетические заболевания, в том числе ФКУ. До появления этого анализа 1 % всех случаев умственной отсталости в США связывали с ФКУ; теперь выявляется всего несколько случаев в год.
К сожалению, не по всем болезням и не во всех штатах проводится такой скрининг. В 2005 году Джим Келли, бывший квотербек в команде «Буффало Биллз» (американский футбол), и его жена Джил потеряли сына Хантера, который скончался от редкого генетического расстройства – болезни Краббе. Подобное заболевание показано в фильме «Масло Лоренцо», оно поддается лечению при условии ранней диагностики. Семья Келли основала фонд «Надежда Хантера», позволивший собрать миллионы долларов на исследование болезни Краббе и более полный скрининг новорожденных. Однако в масштабах такого огромного государства, как США, лишь в Нью-Йорке и еще нескольких штатах младенцев обязательно проверяют на болезнь Краббе, что является весьма постыдным фактом.
Через пятьдесят лет после появления теста забора крови из пятки у новорожденных Роберт Грин и его коллеги из Гарвардской медицинской школы запустили Baby Seq – рандомизированное клиническое исследование, в рамках которого планируется отсеквенировать геномы более сотни новорожденных и отследить в них 1700 заболеваний, начинающихся в детском возрасте. Для Гарварда это всего лишь небольшое клиническое исследование, но оно даст возможность совершить огромный скачок в оценке пользы от всеобщего скрининга новорожденных.
На 1950-е годы пришлось активное развитие цитогенетики – изучения хромосом под микроскопом. В диагностической практике этот подход показал, что при нарушении числа хромосом, когда их больше или меньше нормы, неизбежно возникают тяжелейшие болезни. Проблемы связаны с дисбалансом числа генов, то есть с отклонением от нормы «по два гена от каждого». Подобные расстройства не передаются по наследству, как миодистрофия Дюшенна или муковисцидоз, но все равно являются по сути генетическими; они возникают спонтанно при сбоях в клеточном делении и при образовании дефектных сперматозоидов и яйцеклеток.
Наиболее известная из таких болезней – синдром Дауна. Названа болезнь в честь Джона Лэнгдона Дауна, который в 1866 году впервые описал его характерные клинические признаки. «…Это [внешнее сходство] столь выраженное, что, если посадить таких детей бок о бок, сложно поверить, что они из разных семей». Девяносто лет спустя французский врач Жером Лежен обнаружил, что у детей с синдромом Дауна по три экземпляра одной хромосомы (впоследствии выяснилось, что лишняя 21-я хромосома). Среди генетиков-профессионалов такое расстройство именуется «трисомия 21».
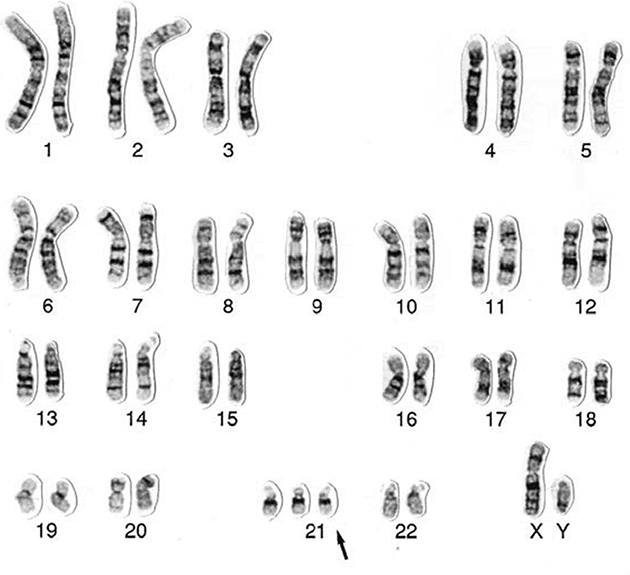
Трисомия 21 (синдром Дауна). Кариотип (полный набор хромосом), взятый у мужчины с синдромом Дауна. Обратите внимание на лишнюю 21-ю хромосому
Чем старше мать, тем выше риск синдрома Дауна. Когда матери 20, вероятность родить такого ребенка составляет примерно 1:1700, когда 35 – уже 1:400, а в 45 достигает 1:30. Именно поэтому многие старородящие женщины выбирают пренатальную диагностику, позволяющую определить, есть ли у плода тройная 21-я хромосома. Сегодня такой анализ входит в стандарт обследования беременных женщин старше 35 лет.
В Великобритании 30 % беременностей с синдромом Дауна обнаруживается при плановом тестировании у 5 % самых старородящих женщин. Этот метод выделяется своей высокой эффективностью по соотношению «число обнаруженных случаев на каждый потраченный фунт стерлингов», но что насчет остальных 70 % случаев синдрома Дауна? Неинвазивные методы являются альтернативными умеренно рискованным технологиям: амниоцентезу и биопсии хориона, – меняют профиль пренатальной диагностики. В конце 1990-х годов различные исследователи, в частности Деннис Ло из Китайского университета в Гонконге, показали, что ДНК плода можно обнаружить в плазме крови матери. Десять лет спустя Ло и группа ученых из Стэнфордского университета под руководством Стивена Квейка независимо показали, что анализ такой ДНК позволяет выявить трисомию 21. Процедура под названием «неинвазивное пренатальное тестирование» (NIPT) относительно проста: исследователь секвенирует 5–10 миллионов коротких произвольно взятых фрагментов ДНК из материнской плазмы, а затем сличает их с соответствующими хромосомами. Если в ДНК плода обнаруживается лишняя 21-я хромосома, то из 21-й хромосомы будет больше фрагментов, чем из других. Аналогично обнаруживаются трисомии и в других хромосомах, в частности в 13-й или 18-й. Такие трисомии вызывают соответственно синдром Эдвардса и синдром Патау, тяжелые генетические расстройства; ребенок с такими болезнями обычно умирает спустя несколько недель или месяцев после рождения. Другие трисомии летальны на пренатальном этапе, так прерывается около 30 % беременностей, и около половины подобных случаев прерывания беременности связаны с теми или иными хромосомными аберрациями.
Точность и эффективность метода неинвазивного пренатального тестирования (NIPT) была доказана в ходе клинических исследований, и первый клинический анализ по такому методу диагностики был выставлен в 2011 году. Вскоре появились и другие, и NIPT стал стандартной диагностической пренатальной процедурой, которую обычно проходят на первых 10 неделях беременности. Увы, этот метод не абсолютно точен. В 2014 году в Атланте был зафиксирован первый «ложноотрицательный» результат NIPT: ребенок родился с синдромом Дауна, хотя диагностика показала, что плод здоров.
Пока скрининг ДНК плода, взятой из плазмы материнской крови, позволяет выявить лишь ограниченный набор генетических расстройств.

Флуоресцентное окрашивание, позволяющее определить число хромосом. Ядро клетки (синее) анализируется на присутствие 10-й хромосомы (голубая) и 21-й хромосомы (розовая). Слева показан нормальный кариотип, в котором по два экземпляра обеих этих хромосом; справа – кариотип, соответствующий синдрому Дауна (три 21-х хромосомы)

Симон – итальянский студент, страдающий синдромом Дауна. Снимок предоставлен Positive Exposure – некоммерческой организацией, цель которой – поддержка «особенных» людей, стремящихся вести нормальную жизнь
Однако, по мнению Джея Шендью (Jay Shendure), врача-исследователя из Вашингтонского университета, вскоре появится возможность считывать по такой ДНК всю геномную последовательность плода. Джей Шендью с коллегами предложили метод неинвазивного взятия образцов материнской ДНК и сравнения этого материала с геномными последовательностями родителей. Так с высокой точностью удается определить геномную последовательность плода. Проблемы остаются, но, как заявил Джей Шендью в беседе с журналистом New York Times, «…это уже не научная фантастика».
По мере того как генетические анализы становятся все более сложными, открывается ящик Пандоры с этическими дилеммами. Подоплека их далеко не ограничивается исходными проблемами, для решения которых разрабатывался анализ, а последствия касаются не только тех людей, которые такой анализ сдают. Ни в какой другой отрасли медицины эта проблема не проявляется так очевидно, как при генетических анализах в семьях с заболеваниями, передающимися по наследству, например миодистрофией Дюшенна, болезнью Хантингтона или муковисцидозом. Недавно мужчина в возрасте за двадцать попросил проверить его на болезнь Хантингтона. Его дед по отцу умер от этой болезни, а отец, которому было уже за сорок, решил не сдавать анализ, предпочтя, подобно Нэнси Векслер, жить с пятидесятипроцентной неопределенностью, а не знать наверняка. Поскольку болезнь Хантингтона обычно поражает пожилых, существовала вероятность, что и у отца есть такая мутация, просто симптомы еще не проявились. Молодой человек понимал, что у него вероятность такой мутации (и, соответственно, вероятность заболеть в будущем) составляет 1 к 4. Но он хотел знать, что его ждет. Проблема состоит вот в чем: если мутацию у сына обнаружат, то получить ее он мог только от отца, и, следовательно, отца болезнь неизбежно настигнет. Стремление сына доискаться генетической правды прямо противоречит нежеланию отца ее знать. Отец с сыном рассорились, и в конце концов, после долгих уговоров матери, сын отказался от анализа. Мать убедила сына в том, что его желание знать правду меркнет по сравнению с правом отца защититься от информации, которая может стать для того смертным приговором. Этот драматический пример показывает, как сильно генетический анализ отличается от любого иного. Любая информация, которую я могу узнать о моих генах, отразится на моих биологических родственниках независимо от того, хотят ли они ее знать или предпочитают скрыть.
Иногда такие следствия могут быть актуальны не для самих носителей мутации, а для их потомков. Синдром фрагильной X-хромосомы – самое распространенное наследственное заболевание, приводящее к умственной отсталости. Кроме сниженного IQ, для больных этим синдромом характерно продолговатое лицо с выдающейся нижней челюстью и оттопыренными ушами, а также гиперактивность и раздражительность. Как и миодистрофия Дюшенна, это расстройство сцеплено с X-хромосомой, но в отличие от миодистрофии Дюшенна поражает и женщин, и мужчин. Очевидно, одной нормальной копии гена недостаточно, чтобы полностью устранить эффект мутантного гена. Тем не менее у женщин эта болезнь протекает легче, чем у мужчин, а частота ее составляет 1:6000 у девочек против 1:4000 у мальчиков. Синдром фрагильной X-хромосомы обусловлен примерно такой же мутацией, как и болезнь Хантингтона: в ДНК повторяется триплет ЦГГ, и эти последовательности растягиваются, как гармошка, достигая патологической длины. У здорового человека от пяти до сорока экземпляров такого триплета, а у больного с синдромом ломкой X-хромосомы – двести и более. По непонятным причинам число таких повторов обычно увеличивается из поколения в поколение, и как только накапливается примерно 230 триплетов ЦГГ, ген больше не может синтезировать тРНК и, соответственно, перестает работать. Болезнь названа по характерной структурной непрочности X-хромосомы, вызванной всеми этими повторами.
По мере увеличения числа повторов из поколения в поколение заболевание становится все тяжелее, болезнь прогрессирует все раньше (этот феномен называется «антиципация»). Последние представители в роду, пораженном синдромом фрагильной X-хромосомы, обычно имеют максимальное число повторов и заболевают раньше, чем предки, от которых им досталась эта мутация. Следовательно, генетики могут выявить индивидов, обладающих «премутацией»: у них не так много повторов, чтобы это отразилось на здоровье, но достаточно, чтобы у их потомков развился синдром ломкой X-хромосомы, поскольку имеющаяся у носителя последовательность, вероятно, должна увеличиться. До сих пор неизвестно, как именно работает белок, кодируемый этим геном, но, по-видимому, он участвует в управлении трансляцией тРНК в белок в синапсах во время онтогенеза.
Я, будучи первым директором проекта «Геном человека», обеспечил финансирование исследований, которые помогают понять, как именно (к добру или к худу) повлияют на жизни миллионов людей те знания, что мы вскоре будем непрерывно получать при помощи секвенаторов ДНК. Исходно я выделил на это 3 % нашего суммарного бюджета (впоследствии эта цифра составила 5 %) и назначил Нэнси Векслер, носительницу болезни Хантингтона, главой совета ELSI, которой было поручено изучать этические, юридические и социальные последствия наших исследований. Одной из первых инициатив ELSI стал ряд пилотных исследований по генетическому скринингу. Во времена, когда каждого новорожденного проверяют на предрасположенность к ФКУ, необходимо задаться вопросом, может ли медицина по каким-нибудь причинам не дать возможность человеку проверить себя на предрасположенность к муковисцидозу, миодистрофии Дюшенна, синдрому ломкой X-хромосомы и другим тяжелейшим человеческим заболеваниям, предсказать которые наука в состоянии. Такие вопросы мы ставили перед собой в начале 1990-х годов. Я думаю, что сегодня мы вседалеко ушли от пилотной стадии, несмотря на появление новых методов скрининга, в частности NIPT.
Анализ на наличие дефектного гена, ответственного за миодистрофию Дюшенна и болезнь Хантингтона, обычно проводился лишь в тех семьях, где уже был хотя бы один больной. Такая практика применяется потому, что эти заболевания редкие, а исследования дорогие. Подобная социальная бухгалтерия – вещь спорная, однако данный аргумент абсолютно несостоятелен в случае муковисцидоза, анализ на который тоже применяется ограниченно. Причины этого мне не ясны. Как вы помните, муковисцидоз встречается с частотой 1:2500 – это одно из самых распространенных генетических заболеваний. Преодолены технические сложности, связанные с тестированием муковисцидоза, который может возникать почти из-за двух тысяч мутаций. Качество новых методов секвенирования повышается одновременно с их доступностью, чем открываются новые возможности для популяционного скрининга. И самое главное, прогресс в лечении муковисцидоза значительно увеличил ожидаемую продолжительность жизни больных; выглядит многообещающей молекулярная терапия, направленная против конкретных мутаций, вызывающих эту болезнь.
Несмотря на удручающее нежелание использовать все преимущества крупномасштабного генетического скрининга, в краткой истории этого метода уже найдутся наглядные примеры крайне успешных скрининговых программ в популяциях, где высок риск различных генетических расстройств.
Гемоглобинопатии – это болезни, вызванные теми или иными нарушениями в работе молекулы гемоглобина. К ним относятся, в частности, различные талассемии и серповидноклеточная анемия; считается, что это самый обширный класс генетических заболеваний; от 5 до 7 % всего населения Земли обладают мутациями такого рода. Как мы уже знаем, ген серповидноклеточной анемии поддерживался естественным отбором, поскольку его наличие сдерживает заболеваемость малярией. Ранее говорилось, что особенно часто гемоглобинопатии встречаются в регионах, эндемичных по малярии. Аналогичные адаптивные преимущества объясняют закономерности распространения других гемоглобинопатий в регионах Африки и Средиземноморья. Некоторые мутации более характерны для конкретных этнических групп Африки и Средиземноморья, где бы сейчас ни жили их представители.
Так, целых 17 % представителей диаспоры греков-киприотов, проживающих в Лондоне, носители талассемий. При тяжелой форме заболевания у человека образуются неполноценные эритроциты, поэтому у больного увеличиваются печень и селезенка, и он рискует не дожить до совершеннолетия. Когда в 1974 году Бернадетт Модел из Медицинской школы Университетского колледжа Лондона запустила программу по систематическому скринингу на предмет этого заболевания, лондонская киприотская община отнеслась к этому с энтузиазмом – киприоты как никто другой понимали всю серьезность этого недуга, который с давних времен так пагубно отражался на их жизни. Аналогичная программа на острове Сардиния позволила радикально снизить частоту талассемий – с 1:250 до 1:4000.
Евреи-ашкеназы – еще одна группа, на себе испытавшая, какими бедами чревата смертельно опасная мутация в изолированной общине. Болезнь Тея – Сакса – жуткий недуг, который встречается у ашкеназов в сто раз чаще, чем в большинстве нееврейских популяций. Дети с болезнью Тея – Сакса рождаются на вид здоровыми, но постепенно развитие ребенка затормаживается, и он слепнет. Примерно в двухлетнем возрасте его начинают мучить судороги. Деградация продолжается до тех пор, пока ребенок не умирает примерно в четырехлетнем возрасте, слепой и парализованный. Остается загадкой, почему болезнь Тея – Сакса так часто встречается среди ашкеназов. Возможно, все дело в том, что они сначала миновали генетическое «бутылочное горлышко» (оценочно около IX–X века), а затем дрейф генов оказал влияние на генофонд относительно небольшой группы евреев, которые откололись от основного этноса и дали начало ашкеназам во времена Второго рассеяния. Аналогичный феномен объясняет, почему такая мутация аномально часто встречается среди франкоканадцев в Квебеке и среди каджунов в штате Луизиана; эта смертоносная мутация скорее всего могла присутствовать в небольшой группе отцов-основателей этих популяций. Альтернативное объяснение связано с преимуществом гетерозигот: так, носитель рецессивного гена болезни Тея – Сакса может обладать повышенной устойчивостью к туберкулезу. Возможно, это преимущество особенно пригодилось европейским евреям, исторически селившимся в густонаселенных центральных городских кварталах, где свирепствовало это заболевание.
Причина болезни Тея – Сакса была обнаружена в 1968 году, когда выяснилось, что эритроциты больных переполнены ганглиозидом GM2. Это вещество – важнейший компонент клеточной мембраны, и у здорового человека избыточный ганглиозид расщепляется за счет синтеза фермента гексозоаминидазы A – химического катализатора-посредника, находящегося в лизосомах и принимающего участие в утилизации ганглиозидов в ЦНС. В 1985 году Рэйчел Майеровиц вместе с коллегами из Национальных институтов здравоохранения удалось выделить ген, кодирующий фермент-гексозоаминидазу A, и показать, что у пациентов с болезнью Тея – Сакса он действительно отсутствует из-за генетического дефекта, вызываемого мутацией гена HEXA, ответственного за синтез этого фермента.
При положительном диагнозе в ходе пренатального скрининга остается единственный выход: прерывание беременности, а среди ортодоксальных евреев-ашкеназов аборты запрещены. К счастью, можно обследовать потенциальных родителей. Поскольку такое решение было морально приемлемо, именно эта программа и была развернута для семейных пар. Четверо из десятерых детей у рабби Джозефа Экштейна из Нью-Йорка умерли от болезни Тея – Сакса прямо у него на глазах. В 1983 году он основал программу Дор Йешорим, «род правых», в рамках которой представители местной ортодоксальной еврейской общины делают тест на болезнь Тея – Сакса. Молодых людей мотивируют бесплатно сдавать такой анализ в школах и колледжах в специально назначаемые дни. Программа исключительно конфиденциальна; даже самому тестируемому не сообщают, является ли он носителем гена; по результатам анализа он получает лишь кодовый номер. Если впоследствии двое молодых людей подумывают вступить в брак, они связываются с Дор Йешорим и называют свои номера. В том случае, если оба потенциальных супруга являются носителями гена, им об этом сообщают и предлагают прийти на консультацию. Такое разглашение по принципу «действительной необходимости» практикуется для того, чтобы носители гена не становились изгоями, однако угрозу болезни Тея – Сакса можно было бы предотвратить.
К настоящему времени в рамках программы Дор Йешорим проверено более 200 тысяч человек и обнаружены сотни пар, относящихся к группе риска, такие мероприятия привели к тому, что сейчас в Северной Америке почти не рождается детей с этой болезнью. Тем не менее некоторые усматривают принуждение в сути самой этой программы: всех молодых людей вынуждают сдавать анализ, а строгие рекомендации многим кажутся столь угрожающими, что некоторые пары даже решают не вступать в брак. Оппоненты заклеймили инициативу рабби Экштейна как «евгенику» (хотя нигде и никогда это слово не имеет такой порочной коннотации, как в еврейском сообществе), однако подобная демагогия едва ли меняет ключевой факт: эта программа действительно пользуется активной поддержкой в той целевой аудитории, для которой предназначена. Реализованная программа Дор Йешорим продемонстрировала, что, с одной стороны, можно уважать культуру, а с другой – успешно участвовать в медицинском скрининге, позволяя программе работать даже в такой ситуации, когда обычаи и религиозные каноны кажутся совершенно несовместимыми с генетическими анализами.
Пренатальный скрининг оборачивается жестким выбором для любой беременной женщины, которая получила положительный результат после проверки на генетическое заболевание: прерывать или не прерывать? Амниоцентез обычно невозможен, пока плоду нет пятнадцати недель, и поэтому прерывание беременности на поздних сроках получается еще более травматичным. На данном этапе при аборте уничтожается не ровненький многоклеточный шарик, а миниатюрное существо, которое вполне можно рассмотреть на УЗИ. Для многих родителей было бы бесконечно предпочтительнее принимать такие тяжелые решения по итогам генетического анализа на более ранних этапах развития плода. Именно поэтому была изобретена преимплантационная генетическая диагностика (ПГД).
Роберт Уинстон, профессор естественных наук и социологии в Имперском колледже Лондона, является ведущим гинекологом-микрохирургом и одним из самых известных на британском телевидении популяризатором науки и биомедицинских исследований. Будучи исключительно разносторонним человеком, он находит время (как лорд Уинстон Хаммерсмитский) заседать в парламенте и консультировать правительство по вопросам его компетенции. Комбинируя две ультрасовременные технологии – экстракорпоральное оплодоторение (ЭКО) и ДНК-диагностику на основе реакции ПЦР, – Роберт Уинстон первым применил метод, позволяющий проверить генетическое состояние эмбриона еще до того, как он прикрепится к стенке матки. После ЭКО несколько продуктов зачатия выращиваются в чашке Петри, пока каждая оплодотворенная яйцеклетка не поделится три-четыре раза и не превратится в шарик из восьми-шестнадцати клеток. У каждого эмбриона аккуратно берутся одна-две клетки, так называемые бластомеры, и из них извлекается ДНК, которая затем при помощи ПЦР-реакции используется для амплификации нужной генетической последовательности и проверки, есть ли в геноме искомая мутация. После этого родителям могут подсаживать только те эмбрионы, проверка которых не выявила генетических заболеваний.
В ходе первых ПГД-анализов, выполнявшихся в 1989 году, проверяли только пол плода – это важная информация, поскольку существует риск генетических расстройств, сцепленных с полом, к которым относится, например, миодистрофия Дюшенна. Так, мать, являющаяся носительницей этого гена, может выбирать лишь эмбрионов-девочек, учитывая, что их болезнь не затронет, хотя они и могут быть ее носительницами. Коллега Уинстона Алан Хендисайд и другие впоследствии расширили область применения ПГД, научившись находить при помощи таких анализов конкретные мутации. В 1992 году этот метод был впервые применен для скрининга муковисцидоза, генетической болезни, не сцепленной с полом.
Поскольку синдром ломкой X-хромосомы может поражать как мальчиков, так и девочек, данное расстройство, естественно, рассматривается в качестве следующей цели для преимплантационной генетической диагностики. Однако исстрадавшимся родителям, знающим, как тяжело растить ребенка с такой болезнью, все равно потребовалось применить все свое влияние, чтобы врачи наконец взялись за решение этой задачи. У Дебби Стивенсон, ранее работавшей продюсером теленовостей, есть сын Тейлор, которому диагностировали синдром ломкой X-хромосомы лишь после того, как мать родила его младшего брата Джеймса. Хотя Джеймс, по счастью, выиграл в «рулетку 50/50» и оказался здоров, Стивенсоны опасались снова испытывать судьбу и заводить третьего ребенка. Тогда они решили попробовать метод ПГД. «Некоторые считают, что неэтично выбирать только здоровые эмбрионы, – говорит Дебби Стивенсон, – но я считаю, что это лучше, чем принимать душераздирающее решение о прерывании беременности, когда уже точно знаешь, что твой ребенок родится тяжело больным». Сейчас в семье Стивенсонов трое здоровых детей, не унаследовавших разрушительной болезни Тейлора.
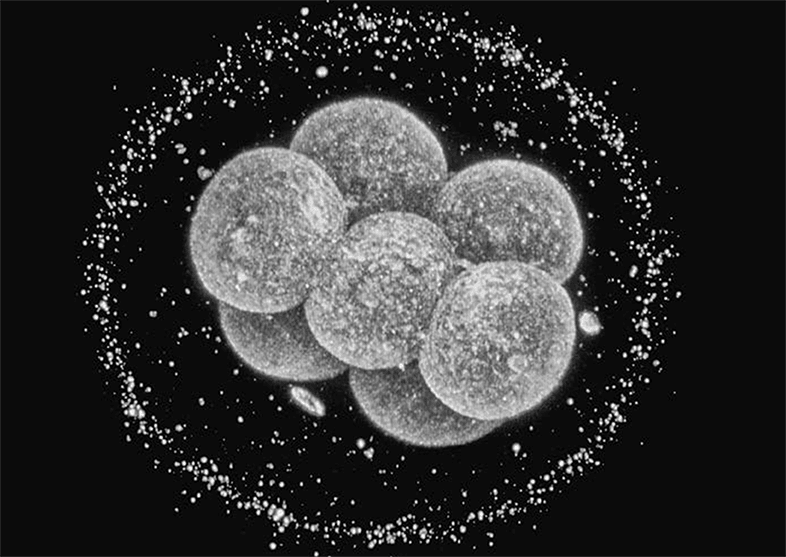
Восьмиклеточный эмбрион
Репродуктивная биология человека кажется неисчерпаемым источником противоречий, а тема, подразумевающая манипуляции над человеческими эмбрионами, по определению взрывоопасна. Преимплантационная генетическая диагностика – не исключение. Тем не менее в последние годы область ее применения расширилась, охватив тысячи редких патологических генов, метод полностью доказал свой потенциал мощного орудия для борьбы с генетическими болезнями. Генетик Марк Хьюз является основателем детройтской клиники под названием Genesis Genetics, предлагающей анализы по всем известным менделевским моногенным заболеваниям, и в частности по болезни Тея – Сакса, миодистрофии Дюшенна, болезни Хантингтона и тысячам других. Кто-то считает, что Хьюз играет с огнем, предлагая ПГД по патологическим генам, которые включаются лишь в зрелом возрасте: ген BRCA1, гены, вызывающие болезнь Альцгеймера с ранним началом, гены, связанные с раком прямой кишки. На замечания и выпады Марк Хьюз говорит: «Я стремлюсь наладить работу диагностической лаборатории, а не руководить курсом по этике. Мне не нужно быть полисменом… Оказывается, люди весьма здраво рассуждают о том, зачем им терпеть такие мытарства, чтобы завести ребенка». Хотя Марк Хьюз вспоминает одну пару, где оба супруга страдали наследственной глухотой. Они заказали у Хьюза ПГД, но хотели не здорового ребенка, а такого же глухого, как и они сами. Хьюз отказал им в диагностических мероприятиях.

Семья Дебби Стивенсон. Старший сын Тейлор болен синдромом фрагильной X-хромосомы. Преимплантационная диагностика показала, что малышка Саманта не унаследует этой болезни
Другая область, в которой эффект ПГД оказался столь же примечательным, но не менее противоречивым, – это работа с так называемымидетьми-спасителями. В августе 2000 года Лиза и Джек Нэш радовались рождению сына, Адама. Перед этим они прошли ПГД, поскольку ранее их единственная дочь Молли родилась больной. Она страдала анемией Фанкони. Адам не только был здоров и не имел анемии Фанкони, его эмбрион был выбран на основе того, что его костный мозг идеально подходил Молли по системе главного комплекса гистосовместимости и в случае необходимости Адам мог бы стать для сестры донором этой ткани и спасти ей жизнь. В Великобритании и в других странах идея подобрать эмбрион как источник «запчастей» для уже имеющегося ребенка многим кажется аморальной. Вопрос спорный, поскольку потенциальное излечение для больного брата или сестры – это как раз тот результат, о котором можно только мечтать.
Значение ПГД трудно переоценить, особенно если семья подвержена генетической болезни из поколения в поколение. Как быть, если таких болезней в семейной истории нет? В 2007 году Энн Моррисс и ее друг, профессор из Гарвардской бизнес-школы, решили создать семью. Они выбрали в банке спермы биоматериал от донора с чертами, которые их устраивали, – так, Моррисс хотела человека с хорошим чувством юмора. Однако в банке спермы почти не дают информации о генетическом скрининге доноров. Исключают только такие факторы риска, как муковисцидоз, серповидноклеточная анемия и болезнь Тея – Сакса. Беременность протекала хорошо, но вскоре после рождения сына Алека Моррисс позвонил врач и тревожно спросил, каково здоровье ее сына, сообщив, что ему угрожает смертельная опасность. Скрининг новорожденного показал, что у Алека редкое наследственное рецессивное заболевание, встречающееся примерно 1 раз на 17 тысяч человек: недостаточность среднецепочечной дегидрогеназы жирных кислот (MCAD). Несмотря на всю ничтожность такого шанса, Адек унаследовал мутантные гены и от Энн (у которой в семейном анамнезе не было случаев этой болезни), и от анонимного донора спермы. Ген кодирует фермент под названием «среднецепочечная дегидрогеназа жирных кислот», обеспечивающий расщепление жиров и извлечение из них энергии. К счастью, результаты поступили в больницу вовремя, еще до того, как у Алека начались какие-либо физические проблемы. С болезнью можно справиться, если пациент с самого раннего детства будет принимать пищу регулярно – раз в пару часов. Этот жизненный опыт одновременно и потряс, и разбередил душу Энн.

Энн Моррисс, генеральный директор компании GenePeeks, делает ставку на потенциал виртуального потомства
Энн Моррисс вместе с генетиком Ли Силвером из Принстонского университета основали диагностическую компанию Gene Peeks, цель которой – сообщать потенциальным родителям генетическую информацию об их будущих, еще не зачатых детях. Алгоритм, разработанный Ли Силвером, моделирует «виртуальное потомство», совмещая на компьютере ДНК двух человек. Примерно за тысячу долларов GenePeeks может спрогнозировать, в каких парах существует повышенный риск рождения детей с какими-либо из тысячи генетических заболеваний. Зная результаты, будущие родители могут воспользоваться ПГД либо взять сперму или яйцеклетки у доноров. Ли Силвер всерьез заявляет: «Предвижу, что в будущем люди будут размножаться без секса – это слишком опасный для размножения метод».
Все расстройства, которые мы здесь рассмотрели, «просты» с генетической точки зрения: они возникают из-за мутации в единственном гене, и ваши шансы заболеть подобным недугом никак не зависят от условий окружающей среды. Выше мы обсуждали рак груди – болезнь, где основной эффект дают конкретные варианты генов, независимо от внешних инициирующих факторов. Мало найдется расстройств, вселяющих такой ужас, как болезнь Альцгеймера, каждый год выхватывающая из жизни все больше людей, которых ждет чудовищная физическая и умственная немощь. Этим синдромом страдают более пяти миллионов американцев. На поздних стадиях болезни человек не понимает, кто он и где находится, не узнает даже ближайших родственников. Поскольку болезнь неотвратимо разрушает память и самосознание, человек разрушается как личность.
Как правило, первые признаки болезни Альцгеймера проявляются в возрасте около 60 лет, но иногда (5 % всех случаев) болезнь приходит уже после сорока. В 1995 году были обнаружены три гена, вызывающих болезнь Альцгеймера с ранней манифестацией. Все они так или иначе связаны с переработкой запасов белка амилоида, который накапливается в мозге у больного; это еще в 1906 году заметил доктор Алоис Альцгеймер, впервые описавший болезнь. На сегодняшний день совершенно очевидно, что болезнь Альцгеймера с ранним началом явно передается по наследству. А что известно насчет более распространенной разновидности этой патологии?
Покойный Аллен Роузес из Университета Дьюка исследовал гораздо более знакомую врачам и распространенную форму болезни Альцгеймера с поздним началом, которая иногда также встречается в разных поколениях одной и той же семьи. Так, Рональд Рейган, объявивший о своей болезнив 1994 году и умерший десять лет спустя, также потерял мать и брата Нила, скончавшихся от болезни Альцгеймера с поздним началом. Аллен Роузес, получивший квалификацию невролога, приступил к поискам в 1984 году. В 1990 году он заявил, что один из генов 19-й хромосомы, по-видимому, коррелирует с болезнью, но его слова восприняли скептически. Однако ничто так не поддталкивало Роузеса к продолжению работы, как возможность доказать неправоту оппонента. Два года спустя он действительно идентифицировал критически важный ген, кодирующий аполипопротеин Е – белок, связанный с переработкой холестерина. Существуют три формы (аллеля) гена: APOE2, APOE3 и, самая важная, APOE4 – даже если у человека всего одна копия такого аллеля, вероятность получить болезнь Альцгеймера увеличивается в четыре раза. Человек с двумя копиями в 10 раз сильнее подвержен риску, чем люди, вообще не имеющие аллеля APOE4. Аллен Роузес обнаружил, что у 55 % людей с двумя копиями аллеля APOE4 к 80 годам развивается болезнь Альцгеймера. При этом у множества людей, обладающих двумя копиями APOE4, болезнь Альцгеймера не случается. Однако скрининг на APOE4 в комбинации с клиническими обследованиями действительно повышает точность диагностики болезни Альцгеймера. В главе 8 я упоминал, что, поскольку ген APOE4 как таковой не является предвестником болезни Альцгеймера, для себя я не пытался выяснить, сколько у меня копий этого аллеля. Поэтому мой статус относительно гена APOE4, в отличие от всех остальных генов в моем геноме, остается абсолютной тайной для всех – в том числе и для меня.
Что же насчет лечения большинства генетических заболеваний? Мы уже достаточно знаем о многих из них, чтобы проводить раннюю диагностику, возможно, избегать, но почти не умеем их лечить. К счастью, в отдельных случаях наши генетические поиски помогли победить болезнь до конца и подобрать для некоторых заболеваний эффективную терапию.
Одна из наиболее воодушевляющих и свежих историй: борьба с распространенным заболеванием – высоким уровнем холестерина. По иронии судьбы этот этап связан с поиском редких мутаций у человека и, можно сказать, является побочным продуктом поиска. «Из всех интригующих последовательностей ДНК, о которых “проболтался” геном человека, – писал Стивен Холл, – наиболее многообещающей, пожалуй, выглядит мутация гена PCSK9, способная быстро и масштабно повлиять на здоровье человека». Липопротеин низкой плотности, так называемый «плохой» холестерин, выводится из кровотока рецепторами липопротеинов низкой плотности, расположенными на поверхности клеток печени. Такое взаимодействие регулирует белок под названием пропротеиновая конвертаза субтилизин-кексинового типа 9 (PCSK9), имеющий сродство к рецептору липопротеинов низкой плотности. В 2003 году исследователи сообщили, что у пациентов с редкой формой гиперхолестеринемии в гене PCSK9 обнаружена мутация «с приобретением функции»: ген продуцировал слишком много такого белка, блокируя, таким образом, работу рецептора и мешая ему связывать «плохой» холестерин. Хелен Хоббс из Юго-Западного медицинского центра Техасского университета заключила, что снижение уровня рецептора липопротеинов низкой плотности вызывает пониженный метаболизм липопротеинов низкой плотности, что может привести к гиперхолестеринемии. Для проверки этой гипотезы Хелен Хоббс обследовала пациентов, участвовавших в кардиологическом исследовании Dallas Heart Study. Она пришла к выводу, что некоторые мутации гена PCSK9 могут приводить как к увеличению, так и к снижению холестерина крови. Мутации, которые нарушают связывание PCSK9 с липопротеинами низкой плотности, приводят к увеличению эффективности работы рецептора и, как следствие этого, к понижению концентрации холестерина в крови. У таких носителей наблюдается пониженный холестерин липопротеинов низкой плотности и снижение риска инфаркта миокарда и другой сердечной патологии. Наиболее ярко эффект такого рода проявлялся среди афроамериканцев. Более того, она нашла одну здоровую женщину – афроамериканку, тренера по аэробике, которой было уже за сорок, гомозиготную по признаку мутации PCSK9. Белок PCSK9 у нее вообще не образовывался, а уровень холестерина составлял всего 14 мг/дл.
Убедившись, что генетическое типирование с PCSK9 в качестве мишени позволяет радикально снизить «плохой холестерин», фармацевтические компании принялись наперегонки разрабатывать лекарства, которые могли бы работать точно таким же образом. За десять лет было создано и одобрено два препарата направленного механизма действия с использованием моноклональных антител: один произвела компания Amgen, другой был совместным продуктом Regeneron (США) и Sanofi (Франция). Ожидается, что эти и подобные вещества станут фармацевтическими блокбастерами, востребованными не менее, чем статины (группа препаратов, действие которых направлено на снижение уровня холестерина в крови).
Довольно часто генетические заболевания приводят к прогрессирующему отмиранию клеток в той или иной ткани. При миодистрофии Дюшенна это происходит с мышцами, при болезни Хантингтона и Альцгеймера – с нервными клетками. Подобный патологический распад пока предотвратить невозможно. Пока мы в самом начале долгого пути, хотя мне порой кажется вполне вероятным, что со временем мы научимся лечить такие болезни при помощи технологии стволовых клеток. Большинство стволовых клеток в организме способны порождать лишь сами себя: так, из стволовой клетки печени может вырасти лишь полноценная клетка печени, но некоторые из них могут давать различные специализированные типы клеток. Простейший пример – свежеоплодотворенная яйцеклетка; это стволовая клетка с максимальным потенциалом. В итоге из нее образуются клетки всех известных типов, у человека их различают две сотни или более. Соответственно, стволовые клетки удобнее всего получать из эмбрионов; у взрослых они тоже встречаются, но такие стволовые клетки лишены эмбриональной способности превратиться в клетку любого типа. Работа по освоению потенциала этих многофункциональных клеток, по определению, связана с уничтожением эмбрионов. Из-за этических возражений против такой практики появились ограничения на выращивание новых линий стволовых клеток, из-за чего клеточного материала для таких исследований всегда не хватало. Наконец, в мае 2009 года эти ограничения были сняты специальным исполнительным указом по результатам опросов, отражающих волю большинства американцев.
Любой атеист согласится, что случится настоящая трагедия, как для науки, так и для всех, кто мог бы излечиться при помощи стволовых клеток, если исследования в этой области станут тормозиться из-за протестов религиозного характера. Однако дискуссии на морально-этические темы возникают повсеместно, и их исход прогнозировать весьма затруднительно. К счастью, последние исследования показывают, что существуют и альтернативные способы добиться многофункциональности стволовых клеток, не требующие уничтожать эмбрионы. Важнейшее достижение произошло в 2006 году, когда Синья Яманака и его коллеги из Киотского университета в Японии продемонстрировали четыре фактора транскрипции, способные менять закономерности регуляции генов и перепрограммировать клетки взрослого организма, превращая их в стволовые. Всего через шесть лет Яманака был удостоен Нобелевской премии за это открытие. Гарвардский ученый Дуг Мелтон, специализирующийся на биологии развития, посвятил свою карьеру исследованию стволовых клеток после того, как его сыну Сэму диагностировали диабет I типа. В 2014 году группа Мелтона сообщила, что из человеческих эмбриональных стволовых клеток удалось синтезировать β-клетки поджелудочной железы, которые были вживлены мышам и стали в их организме вырабатывать инсулин. СМИ поторопились, возвестив о прорыве в лечении диабета, думаю, потребуются годы клинических исследований, прежде чем эти надежды воплотятся в реальность.
На сегодняшний день лечение генетических заболеваний не вышло на уровень полноценной замены старых клеток на новые, выращенные из стволовых. Тем не менее мы уже умеем обеспечивать человека недостающими белками. Болезнь Гоше – редкое расстройство, встречающееся с частотой примерно 1:50 000. Оно возникает из-за мутации в гене глюкоцереброзидазы, приводящей к его недостаточности, которая в свою очередь приводит к накоплению глюкоцереброзида во многих тканях, включая селезенку, печень, почки, легкие, мозг и костный мозг. В 1994 году биотехнологическая компания Genzyme, воспользовавшись рекомбинантными методами, приступила к производству модифицированной разновидности фермента, который позволил бы обеспечить пациентов с дефектным геном этим жизненно важным белком.
Метод заместительной ферментной терапии не просто осуществим, но и весьма эффективен. Однако лечение обходится дорого, около 300 тысяч долларов в год. В 2014 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) одобрило разработанный Genzyme пероральный препарат для лечения некоторых форм болезни Гоше, но стоимость его остается крайне высокой. Биотехнологические компании отстаивают право высокой цены на препараты, направленные против так называемых орфанных (очень редких) заболеваний, мотивируя это высокой стоимостью этапов разработки лекарств, необходимостью инвестирования в научно-исследовательские и опытно-конструкторские технологии и поддержание бизнеса на устойчивом уровне. Специалисты по патентному праву и некоторые политики считают, что отдельные компании просто «задирают цены», являясь монополистами.
Естественно, генетики давно мечтают разработать методы, которые позволили бы устранить проблему в корне, а не просто смягчить ее эффекты. Идеальное лечение генетических болезней, внесение тех или иных генетических изменений, исправление проблемных генов. Результаты такого лечения будут сохраняться пожизненно: излечив болезнь, мы устраним ее окончательно. Для этого существуют, по крайней мере в принципе, два метода: соматическая генная терапия, при которой мы меняем гены прямо в клетках пациента, и терапия зародышевой линии, когда мы работаем с генами в сперматозоидах и яйцеклетках пациента, не допуская передачи вредных мутаций следующему поколению.
Идея генетической терапии в обществе широкой поддержкой не пользуется. Если общество настороженно относится к генетической модификации кукурузы, можете себе представить, какой отпор встречает идея создания трансгенных людей – генно-модифицированных гуманоидов, если хотите, – при всей ее потенциальной пользе. Также ожидаемо, что еще более громкие претензии выдвигаются по поводу генетического изменения зародышевой линии из-за этических опасений по поводу перманентного изменения нашей генетики и рисков, связанных с повреждением генов при манипуляциях с ДНК. При соматической генетической терапии такой ущерб можно ограничить, но при работе с зародышевой линией, в принципе, возможно случайно повредить жизненно важный ген и породить неполноценного человека. Даже поборники генных методов терапии, именно к таким я себя отношу, никогда не станут выступать за распространение таких процедур, пока не будет доказана их полная безопасность. Многие ученые считают, что за генетическую терапию зародышевой линии вообще не следует браться. До недавних пор такие споры оставались чисто академическими: генетическая терапия зародышевой линии была техническиневозможна. Все изменилось с появлением новой технологии CRISPR, о которой я расскажу чуть ниже. Пока давайте поговорим о злоключениях, связанных с соматической генетической терапией.
Первый бесспорный практический успех генетической терапии относится к 1990 году и является заслугой Френча Андерсона, Майкла Блеза и Кена Кулвера из Национальных институтов здравоохранения. Они решили поработать с очень редким расстройством, именуемым аденозиндезами-назная недостаточность, при котором не образуется фермент аденозиндезаминаза (adenosine aminohydrolase, ADA), что обусловливает тяжелую форму иммунодефицита, тип наследования – аутосомно-рецессивный. Молекула аденозиндезаминазы является бифункциональной и, с одной стороны, участвует в регуляции синтеза предшественников нуклеиновых кислот, а с другой – определяет структуру рецептора интерлейкина-2, важнейшего цитокина, обеспечивающего кооперацию Т- и В-клеток в иммунном ответе. В эксперименте участвовали две маленькие девочки с подобным заболеванием: четырехлетняя Ашанти де Силва и девятилетняя Синди Катшелл.
Как внедрить новый ген в клетки пациента? На тот момент казалось логичным сделать это при помощи ретровирусов. Как правило, эти вирусы сравнительно мягко и нетравматично обращаются с клеткой-хозяином: вирус размножается, не разрушая клетку. При помощи генной инженерии ученые создали ретровирусы, которые максимально безопасны для использования при генетической терапии. У них удалены все гены, кроме тех, что абсолютно необходимы для встраивания в геном клетки-хозяина.
Однако как нацелиться лишь на клетки, затронутые мутацией, те, в которых нужно заменить ген? Для первого, пилотного испытания генетической терапии аденозиндезаминазная недостаточность была выбрана удачно: те иммунные клетки, которые нуждаются в коррекции при этой болезни, легко доступны, так как присутствуют в кровотоке в значительном количестве. Команде Андерсона удалось извлечь из крови девочек миллионы лимфоцитов, культивировать их, затем инфицировать ретровирусом, несущим функциональную копию гена. После инкорпорации в естественную клеточную ДНК гена-заменителя клетки пациента можно было повторно ввести в организм.
В сентябре 1990 года Ашанти де Силва первой прошла эту процедуру, а Синди Катшелл четыре месяца спустя. Обе получали инфузию генетически измененных иммунных клеток раз в несколько месяцев. Параллельно обе продолжали проходить негенетическую терапию по замене ферментов, но в меньших дозах: считалось слишком опасным испытывать на девочках новые методы лечения без такой подстраховки. По-видимому, эксперимент стал работать: иммунитет у обеих улучшился, они стали лучше справляться с инфекциями. Могу лично подтвердить, что Синди выглядела весьма здоровой одиннадцатилетней девочкой, когда в 1992 году вместе с семьей побывала у меня в лаборатории Колд-Спринг-Харбор. Сегодня обе эти женщины здоровы; для своих семей они героини, сыгравшие историческую роль в развитии генетической медицины, пусть даже оба случая слишком неоднозначны, чтобы считать их бесспорным успехом генетической терапии.

Синди Катшелл – одна из первых пациенток, прошедших генетическую терапию. Побывав в лаборатории Колд-Спринг-Харбор, она прислала рисунок, где я был изображен за работой
Клинические испытания с участием Катшелл и де Силва были проведены через десять лет после первого в истории эксперимента по генетической терапии. Этот эксперимент окончился неудачей и породил такие серьезные противоречия, что правительство США убило это новое начинание в зародыше. Мартин Клайн был умным и амбициозным врачом, стремившимся облегчить страдания своих пациентов, особенно тех, кто страдал β-талассемией. После успешных экспериментов на животных Клайн обратился в наблюдательный совет Калифорнийского университета в Лос-Анджелесе (именно в этом университете он и работал), запросив разрешения опробовать на людях генетическую терапию с применением нерекомбинантной ДНК. В то время как его заявка находилась в стадии рассмотрения, нетерпеливый Мартин Клайн договорился о том, чтобы применить свои методы для лечения двух женщин за пределами США (в Израиле и в Италии), при этом воспользовался рекомбинантными генами. Одобрения Национальных институтов здравоохранения США получено не было. Вернувшись в Лос-Анджелес, Клайн узнал, что его просьбу отклонили. И не мудрено. Он нарушил практически все ранее существовавшие правила: без разрешения лечил пациентов и пользовался при этом однозначно запрещенными методами. В результате Мартин Клайн лишился федерального финансирования и был вынужден уйти с поста руководителя своего отдела. Генетическая терапия потеряла своего первого практика.
Случай с Клайном, конечно, оказался не последним, когда ученые, пытавшиеся применять генетическую терапию, действовали наперекор социальным регуляторам. К сожалению, только смерть пациента в ходе клинического испытания оказалась достаточно веским доводом, чтобы донести печальную истину: генетическая терапия опасна, поэтому требуется строгий надзор за всеми процедурами, связанными с лечением людей. Джесси Джелсинджер умер не только из-за того, что наших знаний было недостаточно, чтобы с полной уверенностью спрогнозировать реакцию конкретного организма на генетическую терапию, но и потому, что ученые позволили себе непозволительную служебную небрежность.
В 1999 году Джесси Джелсинджер, юноша из Аризоны, узнал об эксперименте, который проводил Джеймс Уилсон, директор Института генетической терапии при Пенсильванском университете. Джелсинджер страдал недостаточностью орнитинтранскарбамилазы (OTC), наследственным заболеванием, при котором печень не способна перерабатывать мочевину, естественный продукт белкового обмена веществ. Эта болезнь смертельна, если ее не лечить, и, хотя у Джелсинджера была лишь легкая форма, он вызвался добровольцем, надеясь, что с его помощью будет найдено лекарство для себя и для других. В лечебной программе Пенсильванского университета в качестве генотерапевтических векторов использовали аденовирусы (один из вирусов этой группы вызывает простуду). Но через несколько часов после того, как вирусы внедрили нормальный ген орнитинтранскарбамилазы в клетки печени Джелсинджера, у того началась лихорадка. За действиями генетиков последовала крайне тяжелая инфекция, сопровождавшаяся тромбообразованием и печеночными кровотечениями. Через три дня после инъекции Джесси Джелсинджер умер.
Смерть юноши шокировала не только его семью, но и все исследовательское сообщество. Подробный ретроспективный анализ показал, что, хотя ранее в том же исследовании у двух пациентов наблюдались признаки печеночной интоксикации, об этих случаях не сообщили в регулирующие органы, кроме того, скрыли эту информацию от всех добровольцев, участвовавших в исследовании. Трагедия серьезно подкосила развитие генетической терапии. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов временно заблокировало все подобные эксперименты в Пенсильванском университете и где бы то ни было. Президент Клинтон призвал повысить стандарты «информированного согласия», ратуя за право участников исследований знать обо всех потенциальных рисках. Если смерть Джелсинджера и принесла какую-либо пользу, то она состояла в ужесточении федерального надзора за клиническими испытаниями с участием людей.
Генно-терапевтическое сообщество все еще было взбудоражено произошедшими грустными событиями, когда из-за океана пришли вдохновляющие новости о достигнутом успехе. В 2000 году команда под руководством Алейна Фишера в парижской детской больнице Неккер применила генетическую терапию для лечения двух маленьких детей, которые страдали тяжелым комбинированным иммунодефицитом, поэтому с рождения содержались в стерильных камерах. Как и в случае с лечением аденозиндезаминазной недостаточности, врачи использовали ретровирус для доставки необходимого гена в клетки, взятые у малышей; затем эти клетки вводились им повторно. Однако необходимо отметить важную инновацию: французские исследователи собирали клетки для последующей модификации из костного мозга детей, стремясь получить самовоспроизводящиеся генетические заплатки. При делении стволовых клеток увеличивается не только количество самих этих клеток, но и количество специализированных соматических клеток, в которые стволовые клетки естественным образом превращаются при дифференциации. Следовательно, любые Т-лимфоциты, получающиеся из измененных стволовых клеток, также будут нести внедренный ген, поэтому повторные инфузии модифицированных клеток уже не нужны.
Именно так и вышло: десять месяцев спустя у обоих пациентов уже были Т-лимфоциты с рабочей копией ранее отсутствовавшего гена, и их иммунная система работала так же хорошо, как и у обычных детей. С тех пор методом Фишера лечили многих детей, страдающих тяжелым комбинированным иммунодефицитом. После долгого и не самого благоприятного начального этапа генетическая терапия наконец подошла к бесспорному успеху. Однако пить шампанское пришлось недолго. В октябре 2002 года врачи обнаружили у одного из тех первых пролеченных пациентов лейкемию – рак костного мозга. Напрашивался неизбежный вывод: генетическая терапия помогла ребенку излечиться от тяжелого комбинированногоиммунодефицита, но в качестве побочного эффекта дала рак. Таково грустное напоминание о том, что генетическая медицина, как и более традиционные фармацевтические методы, может обернуться непредвиденными последствиями.

Тогда казалось, что все хорошо: Алейн Фишер и Марина Каваццана-Кальво в апреле 2000 года представляют результаты своего фундаментального прорыва в области генетической терапии
Смерть Джесси Джелсинджера и инциденты с лейкемией заострили внимание на множестве сложных проблем, которые предстоит решить для того, чтобы соматическая генетическая терапия превратилась в медицинский мейнстрим. Но эта дисциплина постепенно совершенствуется, уже достигнуты заметные успехи в лечении рака, гемофилии и редкой формы слепоты. Возможно, самым ярким воплощением новой генетической терапии можно считать Кори Хааса, которому в 2008 году сделали операцию на глазах в детской больнице Филадельфии – всего в нескольких милях от той больницы, где ранее умер Джелсинджер. Через четыре дня он громко и пронзительно закричал на входе в филадельфийский зоопарк, когда впервые увидел яркое солнце. Кори страдал редкой формой слепоты, наследуемой по рецессивному принципу, врожденным амаврозом Лебера. Из-за дефектного гена RPE65 или иных генов, кодирующих белки сетчатки, участвующие в восприятии света, в сетчатке умирают и не восстанавливаются светочувствительные клетки. Ген RPE65 был введен Кори при помощи другого вектора, аденоассоциированного вируса, который, судя по всему, безвреден для человека и избирательно внедряет свою ДНК в конкретный участок человеческого генома; таким образом, была сведена к минимуму вероятность повредить какой-нибудь жизненно важный ген.
Однако некоторым сторонникам генетической терапии не терпится наконец-то дождаться реального прогресса. В 2015 году сорокачетырехлетняя Элизабет Пэрриш, генеральный директор биотехнологической компании Bio Viva из Сиэтла, вошла в историю, пройдя неоднозначную генетическую процедуру, в рамках которой попыталась обратить старение вспять. Не получив от Управления по санитарному надзору за качеством пищевых продуктов и медикаментов соответствующего разрешения, Элизабет Пэрриш отправилась в колумбийскую клинику, где ей сделали инъекцию теломеразы, применив в качестве вектора аденоассоциированный вирус. По сообщению Bio Viva, Пэрриш стала «первым человеком в истории, успешно омолодившимся при помощи генетической терапии» – якобы теломеры у Элизабет Пэрриш стали длиннее. В принципе, я считаю, что риск – дело благородное, но опасаюсь, что генетическая терапия может быть вновь отброшена назад, если начинание Пэрриш не будет успешным или приведет к осложнениям.
Современная генетическая терапия далеко не ограничивается доставкой фунциональных последовательностей ДНК для замены поврежденного гена. В 2006 году двое американских ученых, Энди Файр и Крейг Мело, совместно получили Нобелевскую премию за открытие биологического процесса под названием РНК-интерференция (РНКи). РНКи – это естественный механизм, при помощи которого организм обнаруживает и разлагает инородный генетический материал. Если приспособить такой процесс для отбраковки дефективной РНК, продуцируемой мутантными генами, пока она не успела выстроить на основе такого «генетического сценария» нездоровый белок, то открывается захватывающий путь для излечения множества болезней. Несмотря на то что первые клинические испытания РНКи проходят с переменным успехом, некоторые биотехнологические компании, например Alnylam, уже нацелены на борьбу с рядом заболеваний, среди которых гемофилия, гиперхолестеринемия и гепатит.
В дальнейшем генетическая медицина позволит не просто внедрять в организм здоровый ген, но и исправлять или ремонтировать дефектные последовательности в ДНК. Научное сообщество по-настоящему зачаровано одним из таких методов, который позволяет использовать экстраординарный терапевтический потенциал естественного механизма редактирования генов, именуемого CRISPR (короткие палиндромные повторы, регулярно расположенные группами). Эти естественные последовательности в ДНК составляют основу рудиментарной иммунной системы у микробов, но ученые блестяще приспособились использовать их для редактирования любых генов. РНК транскрибируются с локусов CRISPR совместно с ассоциированными с CRISPR белками Cas, что обеспечивают адаптивныйиммунитет за счет комплементарного связывания РНК с нуклеиновыми кислотами чужеродных элементов и последующего разрушения их белками Cas9. Использование методик CRISPR-Cas для направленного редактирования геномов является перспективным направлением в современной генной инженерии. Практически молниеносно лаборатории во всем мире приспособили CRISPR/Cas9 для работы с клетками всевозможных тканей, растительных и животных организмов. Потенциальными реализациями этой техники мы во многом обязаны совместным исследованиям Дженнифер Дудны из Калифорнийского университета в Беркли и Эммануэль Шарпентье из берлинского Института инфекционной биологии им. Макса Планка. В 2014 году они получили многомиллионную премию Breakthrough Prize («Премия за прорыв») за то, что одними из первых распознали самые многообещающие перспективы метода CRISPR. Дальнейшие исследования, проведенные Фэн Чжаном из Массачусетского технологического института, показали, что такая система может редактировать гены и в человеческих клетках, но именно Фэн Чжан первым получил патент на данную технологию.


Высший свет CRISPR: научное сообщество во всем мире признает важный вклад Эммануэль Шарпентье и Дженнифер Дудны (сверху; получают в 2017 году премию в Японии) и Фэн Чжана (снизу), хотя институты, в которых работают эти ученые, являются конкурентами за патентное первенство
В дисциплине, где создание медийной шумихи по поводу всех технологических достижений в порядке вещей, CRISPR задает новые стандарты. В то время как я пишу эти строки, в разгаре серьезная патентная тяжба между двумя университетами, где свои открытия совершили Дженнифер Дудна и Фэн Чжан, а отголоски этой борьбы будут слышны повсюду – от совещательных комнат в биотехнологических компаниях до Шведской академии в Стокгольме. Уже созданы десятки генно-инженерных моделей животных, в которых используется технология CRISPR. Исследователи изменили клетки печени у взрослых мышей, чтобы исправить дефектный ген в модели, где, наряду с другими расстройствами, наблюдалась наследственная тирозинемия. Эксперты считают, что более доступные ткани: глаза, легкие и клетки крови – в недалеком будущем могут стать объектом такой терапии. В конце 2016 года китайские исследователи объявили, что использовали CRISPR в инжиниринге Т-лимфоцитов у пациента, страдающего раком легких, намереваясь усилить иммунный ответ на опухолевые антигены (см. главу 14).
CRISPR может оказаться революционным инструментом для борьбы с такими разрушительными инфекционными заболеваниями, как малярия или лихорадка Зика. Например, инженерия москитного генома в рамках так называемого генетического дрейфа могла бы сделать весь вид бесплодным или блокировать передачу малярийного плазмодия или вируса Зика. Миллионы жизней были бы спасены, но откуда нам знать, на самом ли деле генный дрейф совершенно безопасен? «Существуют определенные риски, связанные с выпуском в природу насекомых, генетически измененных в лаборатории, – считает Энтони Джеймс, исследователь из Калифорнийского университета в Ирвайне, – но я думаю, что бездействие ученых в такой ситуации гораздо опаснее».
Неудивительно, что всевозможные варианты применения CRISPR вызывают глубокие этические вопросы, и прежде всего требуется ответ на самый главный вопрос современной молекулярной биологии: следует ли воспользоваться этим методом для перманентного изменения человеческой зародышевой линии? Я еще вернусь к этой теме в следующей главе.

Близнецы-двойняшки, участвующие в ежегодном фестивале в городе Твинсбург, штат Огайо
Назад: Глава 11 Генетическая дактилоскопия. ДНК в суде
Дальше: Глава 13 Что важнее: наследственность или воспитание?

