Книга: Большая книга упражнений для спины: комплекс «Умный позвоночник»
Назад: Приложение 1. Травма спинного мозга: оптимистические прогнозы
Дальше: Спасительные часы – надежда спинальных хирургов
Апоптоз – отсроченная гибель нервной ткани
В настоящий момент признана концепция первичного и вторичного повреждения нервной ткани.
Травматический агент (смещенные фрагменты позвонков, межпозвонковых дисков, инородное тело) приводит к локальному механическому повреждению, нарушению кровообращения и ишемии ткани спинного мозга. В результате этого формируется некротический очаг первичного повреждения спинного мозга. Некрозу подвергаются как тела нервных, глиальных клеток, так и проводящие волокна спинного мозга. Гибель клеток в очаге связана с чрезмерным механическим или ишемическим повреждением, выдержать которое клеточные мембраны не способны. При этом происходит нарушение энергообеспечения, набухание и распад клетки. Накапливаются продукты распада клеток, биологически активные вещества (цитокины), которые активируют универсальную реакцию тканей на травму – воспаление. Повреждение мозга не останавливается на этом этапе, а продолжается длительное время после первичной травмы, захватывая новые изначально не поврежденные участки спинного мозга. Этот процесс получил название вторичного повреждения.
Среди механизмов вторичного повреждения тканей большое значение придается процессам апоптозной гибели клеток.
Повреждение спинного мозга в типичном случае захватывает несколько сегментов и обычно сопровождается некрозом центрального серого вещества, который в различной степени может распространяться на окружающее белое вещество. Во многих случаях обычно сохраняется неповрежденной часть белого вещества на периферии спинного мозга. Микроскопически в этом участке ткани обнаруживается гиперемия с множеством свободных эритроцитов в разрушенных тканях. Петехиальные кровоизлияния связаны с разрывом сосудов и располагаются, главным образом, в центральном сером веществе. Отек ткани развивается в течение нескольких минут и прогрессирует в первые часы. Эндотелий первоначально не поврежденных капилляров отекает, что нарушает кровообращение сегмента и вызывает дополнительную ишемию. Основным морфологическим проявлением первичного повреждения является некротический очаг, который включает обломки разрушенных клеток и клетки, участвующие в развитии воспаления: в течение нескольких дней область травмы инфильтрируется нейтрофилами, за которыми следуют макрофаги. Мононуклеарные клетки и активированная микроглия (местные фагоцитирующие клетки) постепенно производят удаление клеточного детрита и санацию очага повреждения. Параллельно происходит пролиферация астроцитарной глии. Астроциты появляются в большом количестве на 5–6-е сутки после травмы и замещают погибшие клетки и волокна, формируя глиальный рубец. Этот процесс получил название глиоза.
Микроскопический портрет клетки в состоянии некроза очень характерен. На ранних этапах некроза наблюдается нерезкая конденсация хроматина и деградация цитоплазматических структур, позже происходит разрушение мембран и дезинтеграция клетки.
В результате травмы происходит полный разрыв либо частичное повреждение проводящих волокон спинного мозга. Более уязвимыми являются центрально расположенные волокна. Частичное повреждение аксонов связано с нарушением быстрого тока цитоплазмы по ним и проявляется в виде булавовидных расширений уже через 30 минут после травмы. Подобные изменения аксонов могут подвергаться обратному развитию с восстановлением проводимости по волокну. Полный перерыв аксона проявляется в виде натеков цитоплазмы – аксональных шаров. Они явно появляются через 24 часа после повреждения и указывают на прогрессию изменений. Позднее аксональные шары могут преобразовываться в колбы роста и быть источником регенерации нервных волокон.
Подобные аксональные изменения находят как при ушибе, так и при сдавливании спинного мозга.
Локальное повреждение проводящих волокон вызывает разрушение их миелиновой оболочки – демиелинизацию, дегенерацию и гибель аксона, которые распространяются на протяженные участки спинного мозга. Травматическая демиелинизация наблюдается уже в первые часы после травмы и продолжается длительное время. У некоторых пострадавших, переживших травму, достаточно длительный период времени может наблюдаться обратный процесс в виде частичного восстановления миелина волокон спинного мозга.
Со временем поврежденные клетки удаляются макрофагами и наступает санация травматического очага. Локальное разрушение нервной ткани сопровождается образованием мелких полостей в спинном мозге. Непосредственно область геморрагического некроза замещается множественными кистами, которые пересечены глиально-сосудистыми перемычками. В конечном итоге некротический очаг превращается в глиально-соединительнотканный рубец. Количество соединительной ткани рубца зависит от выраженности дефекта спинного мозга. В рубце выделяют три зоны без четких границ: центральную соединительнотканную, глиально-соединительнотканную, глиальную, которая переходит в неповрежденный спинной мозг. Образование кист связано с потерей клеточных элементов ткани мозга и процессом глиоза. Мелкие полости могут сливаться с образованием посттравматических кист более крупного размера.
В позднем периоде травмы очаг повреждения спинного мозга в типичном случае представлен множественными кистами, выстланными глией, растущими отростками клеток задних рогов и нервных корешков, а также сохраненным в различной степени белым веществом на периферии очага травмы. Сохранившиеся волокна в области повреждения находятся в состоянии демиелинизации и теряют способность к проведению нервных импульсов. На отдалении от очага травмы продолжается валлеровская дегенерация восходящих и нисходящих проводящих путей.
Чаще всего не происходит полного анатомического разрыва ткани спинного мозга. По данным Kakulas B. A., из 354 погибших на месте происшествия с повреждением позвоночника у 139 спинной мозг был внешне не поврежденным. Из 125 больных с травмой позвоночника, которые выжили в течение часов или нескольких суток, спинной мозг был перерван только в 17 случаях.
По различным данным, в 70–90 % случаев спинномозговой травмы сохраняется непрерывность ткани спинного мозга на уровне максимального повреждения.
Как правило, часть волокон в спинном мозге остаются неповрежденными даже при крайне тяжелых травмах позвоночника. Это дает надежду на возможность их сохранения и поддержания тем или иным терапевтическим методом.
Таким образом, локальное повреждение спинного мозга приводит к развитию рубцовых и глиальных изменений спинного мозга в области травмы, локальной и системной демиелинизации и дегенерации проводящих путей. Это проявляется грубой и стойкой неврологической симптоматикой в виде параличей, нарушений чувствительности и вегетативной дисфункции.
Апоптоз в поврежденном спинном мозге
В последнее время наибольшее внимание уделяется механизмам вторичного повреждения нервной ткани. Морфологическое изучение травмированного спинного мозга демонстрирует, что разрушение ткани не ограничивается областью воздействия разрушающей силы, а продолжается во времени, захватывая не поврежденные в момент травмы участки мозга и приводя к образованию большего очага повреждения, чем начальная травма. Другими словами, суммарное число погибших клеток спинного мозга значительно превышает количество разрушенных в момент травмы. Современная концепция патогенеза травматического повреждения спинного мозга рассматривает два основных взаимосвязанных механизма гибели клеток: некроз и апоптоз. Морфологическое изучение поврежденного спинного мозга и поиск путей его восстановления выявили, что оба типа клеточной смерти имеют место и в травмированном спинном мозге.
В настоящее время апоптоз рассматривается как наиболее распространенный тип клеточной смерти при травме.
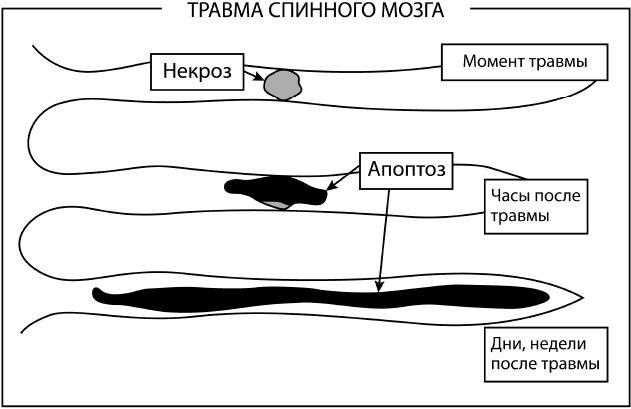
Схема 1. Механизмы повреждения спинного мозга при травме
На схеме 1 представлена общая концепция травматического повреждения спинного мозга.
Изучение апоптоза связано с именем Керра (Kerr J. F., 1972), который ввел этот термин. Длительное время считалось, что апоптоз присущ только эмбриональному развитию. Позднее подобную гибель клеток наблюдали при различных патологических состояниях. Многочисленные исследования последних лет показали, что часто именно апоптоз, а не некроз лежит в основе инфаркта миокарда, острой почечной недостаточности, инсульта, травмы головного мозга и других заболеваний, связанных с высокой смертностью.
Апоптоз наблюдается при разнообразных патологических состояниях центральной нервной системы – травматических, сосудистых, нейродегенеративных болезнях, опухолях.
Открытие апоптоза в поврежденном спинном мозге дало новый толчок к изучению механизмов вторичного повреждения нервной ткани с целью их подавления. Изучение возможности влияния на апоптозную гибель клеток при травме головного или спинного мозга может помочь в лечении и нетравматических заболеваний нервной системы.
Как затормозить апоптоз
Изучение апоптоза при травматическом повреждении спинного мозга является очень перспективным с точки зрения возможности влияния на патологический процесс. В то время как некроз представляет собой необратимую гибель клетки, смерть в результате апоптоза на определенных этапах может быть задержана или предупреждена. Поэтому во многих лабораториях проводятся исследования с целью изучения механизмов активации апоптоза, его временного и пространственного распространения в клеточной популяции ткани. Выясняются индукторы, супрессоры и исполнители программы апоптоза, а также возможные пути влияния на этот процесс, и, прежде всего, его торможения, с целью повышения выживаемости клеток.
Причиной развития апоптоза может быть прямое воздействие на геном клетки (вирусы) или непрямое влияние через нейромедиаторы, медиаторы воспаления, ишемию и прочее. В настоящий момент обнаружено множество факторов, вызывающих апоптоз. Среди них ионизирующее излучение, отсутствие трофических (питательных) факторов, стероидные гормоны, цитокины и прочие. Важную роль в индукции апоптоза при травме спинного мозга отводят возбуждающим нейромедиаторам – глутамату и аспартату, которые в избытке выделяются при повреждении нервной ткани. Значимым для развития апоптоза является накопление универсальных посредников различных биологических процессов – оксида азота (NO), ионов Са2+. Полиэтиологичность апоптоза связывает его со многими патологическими состояниями, такими как травма, ишемия, инфекции.
В неповрежденной клетке процесс апоптоза находится под строгим генетическим контролем. Это связано с тем, что программированная клеточная гибель является необходимым процессом клеточной замены в эмбриогенезе, а у взрослой особи – механизмом естественной элиминации клеток. Апоптозная гибель клетки имеет биохимические критерии, которые отличают ее от процесса некроза. В отличие от него апоптоз – не пассивный, но активный процесс, связанный с активацией генов, регулирующих умирание клетки, который требует энергетических затрат и белкового синтеза. Известно несколько генов, ответственных за развитие апоптоза в спинном мозге. Среди них есть как индукторы – Fas/apo-1, p53, так и ингибиторы апоптоза – bcl-2, bcl-x, bax.
Как и во многих других тканях, апоптоз в спинном мозге представляет собой многостадийный процесс. Были выделены две его стадии: начальная, обратимая фаза апоптоза запускает генетическую программу клеточной гибели, которая заканчивается активацией ДНКаз, ответственных за фрагментацию ДНК. После того, как ДНКазы активированы и началось дробление ДНК, апоптоз переходит во вторую фазу – необратимую, которая заканчивается появлением морфологических признаков апоптоза, дезинтеграцией клетки и ее поглощением макрофагами.
Активация исполнителей апоптоза представляет собой результат разветвленной цепи биохимических реакций, смысл которых состоит в том, чтобы в конечном итоге практически любой внешний повреждающий сигнал мог приводить к фрагментации ДНК и клеточной смерти. Внешний агент – гормон, нейротрансмиттер, цитокин и др. приводят к возрастанию внутриклеточного содержания ионов Са2+. Эти ионы, являющиеся вторичными мессенджерами, поступают в цитоплазму по открывающимся каналам из внешнего пространства и из внутриклеточных депо – митохондрий, эндоплазматической сети. Активный комплекс Са2+-кальмодулин взаимодействует с протеинкиназами, которые активируют исполнителей апоптоза, и в первую очередь – каспазы. Каспазы расщепляют множество белков цитоплазмы, в том числе белки цитоскелета и ядра – фермент репарации ДНК (PARP), гистоновые белки, ингибиторы ДНКаз.
Конечным звеном этого биохимического каскада является активация ДНКаз, которые производят разрушение ДНК клетки. Считается, что 40 двуцепочечных разрывов ДНК на клетку, т. е. примерно 1 разрыв на хромосому, являются летальными. Фрагментация ДНК в клетках с запущенной программой гибели начинается с редких одноцепочечных разрывов ДНК, доходит до частых двуцепочечных разрывов ДНК, достигая размера фрагментов в 200 пар азотистых оснований, что проявляется характерной лестницей при электофорезе. Специфическая фрагментация ДНК является основным критерием для биохимического и иммунногистохимического выявления клеток в состоянии апоптоза.
Динамика апоптоза при травме спинного мозга
Апоптоз в спинном мозге исследуется сравнительно недавно – с середины 1990-х годов. Его изучение проводят при экспериментальном повреждении спинного мозга и предпринимают попытки провести корреляции с процессами, происходящими у человека. Crowe M. J. в 1995 г. представил первые доказательства апоптоза в травмированном спинном мозге крыс. Li G. I. показал, что компрессионная травма спинного мозга связана с апоптозом глиальных клеток, преимущественно локализованных в дегенерированных длинных трактах белого вещества. Именно апоптозная гибель олигодендроцитов является важным фактором вторичного повреждения. Эти клетки продуцируют миелиновую оболочку проводящих волокон, и их апоптозная гибель может быть причиной распространенной демиелинизации и дегенерации волокон. Было выдвинуто предположение об отсроченном апоптозе олигодендроцитов и связанной с ним демиелинизацией после экспериментальной травмы спинного мозга (Blight A. R., 1985) или спинного мозга человека (Bunge R. P., 1993).
До последнего времени единственное описание апоптоза при травме спинного мозга человека было проведено Emery E., которая предприняла первую попытку провести корреляцию между апоптозным повреждением клеток и дегенеративными изменениями в восходящих и нисходящих тактах спинного мозга. В 2000–2003 гг. в совместных исследованиях НИИ нейрохирургии им. Н. Н. Бурденко РАМН и НИИ Мозга РАМН были изучены клинические случаи травмы спинного мозга человека, а также случаи экспериментальной травмы спинного мозга животных с целью изучения динамики апоптоза (Борщенко И. А., Басков А. В., Сатанова Ф. С, Коршунов А. Г.). Исследовались модели травмы в результате пересечения, ушиба и хронической компрессии спинного мозга. Методами иммуногистохимического и биохимического анализа был установлен факт апоптозной гибели клеток спинного мозга крыс как после его экспериментального пересечения, ушиба, так и в случае травматического повреждения спинного мозга человека. При анализе развития апоптоза во времени в случае экспериментального пересечения спинного мозга отмечались два пика этого процесса на 3-и и 40-е сутки после травмы. Данные о двухфазном течении апоптоза в поврежденном спинном мозге подтверждаются другими исследователями. Апоптоз был верифицирован и в глиальных клетках, и в нейронах. Важным является оценка глубины распространения апоптоза от места травмы на протяжении ткани мозга. На всех сроках эксперимента клетки в состоянии апоптоза обнаруживались на протяжении от шейного до поясничного утолщения спинного мозга.
Таким образом, локальное повреждение органа привело к гибели первоначально интактных клеток на протяжении практически всего спинного мозга.
Неустраненная компрессия спинного мозга, в том числе в случае травматического повреждения спинного мозга человека, является причиной длительной и распространенной апоптозной гибели клеток. Эти данные обосновывают максимально ранние декомпрессивные вмешательства на позвоночнике и спинном мозге с целью ограничения и уменьшения процессов вторичного повреждения спинного мозга, и в частности, апоптоза.
Исследование травмированного спинного мозга человека подтвердило данные экспериментальных исследований. Максимальное развитие апоптоза наблюдалось на отдалении от очага травмы. Такое распространение процесса, по-видимому, связано с завершением деструктивных процессов повреждения в зоне первичного повреждения и распространением волны апоптоза вдоль спинного мозга в дистальном и проксимальном направлении. Основными видами клеток, подвергшихся апоптозу, были глиоциты, однако регистрировалась и апоптозная гибель нейронов.
ДНКазы являются основными исполнителями апоптоза, поэтому, суммируя результаты исследования активности ДНКазы в ликворе и в месте поражения, а также на основании иммуногистохимического выявления апоптоза в срезах спинного мозга, можно сделать вывод, что апоптоз у человека начинается в зоне травматического повреждения спинного мозга и распространяется по спинному мозгу на значительное удаление в проксимальном и дистальном направлениях, и максимальная интенсивность процесса наблюдается в первые 2 месяца после травмы.
Назад: Приложение 1. Травма спинного мозга: оптимистические прогнозы
Дальше: Спасительные часы – надежда спинальных хирургов

