Книга: Мусорная ДНК. Путешествие в темную материю генома
Назад: Как провести импринтинг заново
Дальше: Влияние катастрофического события
Когда импринтинг идет не так
Исследования расстройств, связанных с импринтингом, по-настоящему начались в 1980-е годы, когда впервые стала возможной идентификация генов, связываемых с наследственными заболеваниями. Для этого, в частности, отыскивали семьи, где хотя бы один человек страдал от данной болезни, и затем проводили в этих семьях анализ, пытаясь приблизительно выявить область хромосомы, ставшую причиной заболевания. Сегодня мы можем проделывать это довольно легко, ведь у нас есть полная расшифровка нуклеотидной последовательности нормального человеческого генома и доступ к весьма дешевым технологиям секвенирования. Однако тогда, в 1980-е, найти мутацию, вызывающую болезнь, было не так-то просто — все равно, что отыскать определенную перегоревшую лампочку, когда известно лишь, что она перегорела в каком-то американском доме. Для выявления мутаций, причин той или иной болезни, требовались годы упорного труда больших коллективов ученых.
Целый ряд таких научных групп занимался синдромом Прадера-Вилли. Младенцы, родившиеся с этим синдромом, слишком мало весят при появлении на свет, а кроме того, у них нарушен сосательный рефлекс. Лишь после отлучения от груди у них начинает нормально развиваться тонус мышц, а до этого младенческое тельце довольно вялое. По мере взросления у таких детей просыпается ненасытный аппетит. В результате у них рано возникают экстремальные формы ожирения. Кроме того, они страдают от умственной отсталости, пусть и проявляемой в мягкой форме19.
Совершенно другая группа ученых занималась исследованием заболевания с совсем иными симптомами. Речь идет о синдроме Ангельмана. У детей с этим синдромом маленькая, недоразвитая голова, они с трудом обучаются, а кроме того, очень поздно переходят на твердую пищу. Такие дети склонны к беспричинным взрывам смеха. К счастью, отвратительно-бестактное описание этих пациентов как «счастливых манекенов» употребляется сейчас все реже20.
Представьте, что вы прокладываете железную дорогу через весь континент. Одна бригада рабочих начинает с востока и продвигается на запад, а другая идет ей навстречу. Вначале бригады находятся на совершенно различных территориях, однако постепенно сближаются. В конце концов (если все идет как надо), они встречаются в некоей точке, пожимают друг другу руки, выпивают в честь окончания работы. Что-то подобное случилось и с группами, исследовавшими синдромы Прадера-Вилли и Ангельмана. Только вот ученые, в отличие от железнодорожных рабочих из нашего примера, вовсе не ожидали, что встретятся. Они считали, что строят независимые железные дороги в совершенно разные города. И все-таки они очутились в одном и том же месте.
По мере того, как набирало обороты картирование хромосомных зон, ответственных за синдромы Прадера-Вилли и Ангельмана, становилось все яснее, что на эти два заболевания влияет одна и та же область генома. Поначалу выдвигалось наиболее очевидное предположение: причина этих заболеваний — два разных гена, расположенных очень близко друг от друга. Однако в конце концов выяснилось, что оба этих заболевания вызывает дефект на одном и том же строго определенном участке генома.
Оба недуга имеют одну и ту же генетическую подоплеку — утрату небольшого участка хромосомы 15. Родители больных детей не страдали от этих заболеваний. Когда ученые исследовали хромосомы родителей, выяснилось, что эти хромосомы у них не повреждены. Утрата важнейшего участка хромосомы 15 происходила в процессе формирования яйцеклеток или сперматозоидов.
Казалось очень странным, что удаление небольшой части хромосомы способно вызывать два таких разных заболевания. Загадка стала проясняться, когда ученые показали: важно даже не само отсутствие этого маленького участка хромосомы 15. Важно то, почему он отсутствует. Как выяснилось, 70% изученных детей с синдромом Прадера-Вилли унаследовали аномальную хромосому 15 от мутантных клеток сперматозоидов. А 70% детей с синдромом Ангельмана унаследовали аномальную хромосому от мутантных яйцеклеток. Чуть позже исследователи установили, что 25% изученных детей с синдромом Прадера-Вилли обладали двумя совершенно нетронутыми хромосомами 15, в которых не наблюдалось никакой нехватки генетического материала. Дело в том, что эти пациенты наследовали обе копии хромосомы 15 от матери, а не по одной копии от каждого из родителей. Меньшая доля страдающих синдромом Ангельмана имела по две нормальные копии хромосомы 15, причем обе копии наследовались от отца.
Такие картины наследования обретают смысл, только если привлечь концепцию импринтинга (см. рис. 10.3). Во всех аномальных ситуациях в клетках пациента отсутствует контролирующая импринтинг зона, которую ему следовало получить от одного из родителей. Результат — аномальные уровни экспрессии генов, которые в обычных условиях находились бы под жестким «родительским контролем». Это приводит к патологиям, в том числе к недостаточному или чрезмерному развитию органов и тканей.
Исследователи сумели еще больше сузить круг проблем, которые могли бы приводить к этим заболеваниям. Для этого они проанализировали гены, управляемые зонами, контролирующими импринтинг. Выяснилось, что среди обследованных пациентов, страдающих синдромом Ангельмана, примерно 10% унаследовали всю нужную ДНК от каждого из родителей. Однако у них имеется мутация в ДНК, унаследованной от матери. Она происходит не в ОКИ, а в гене, управляемом ОКИ. Это ген, кодирующий белок. Обычно данный ген экспрессируется лишь на хромосоме, наследуемой от матери. На хромосоме, наследуемой от отца, этот ген глушится импринтингом. Если ген, полученный от матери, не в состоянии вырабатывать белок из-за мутации, это означает, что такая клетка вообще не может синтезировать данный белок, что и приводит к патологии.
С синдромом Прадера-Вилли еще более необычная ситуация. Удалось выявить небольшое количество пациентов, у которых отсутствует лишь один из генов, находящихся на этом важнейшем участке хромосомы 15. Этот ген не кодирует белок, однако он кодирует целый набор некодирующих РНК. Все эти РНК обладают сходными функциями21,22,23: они вовлечены в процессы регуляции еще одного класса РНК, не кодирующих белки. Похоже, отсутствие одного-единственного гена, не кодирующего белок, имеет определяющее значения для развития большинства симптомов, характерных для синдрома Прадера-Вилли.
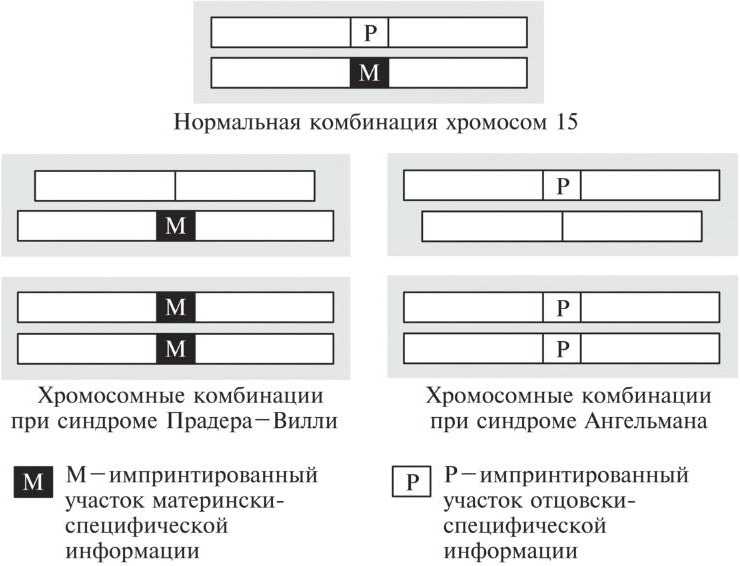
Рис. 10.3. Обычно мы наследуем одну копию хромосомы 15 по материнской линии, а одну — по отцовской. Если обе копии наследуются по материнской линии, у ребенка возникает синдром Прадера-Вилли. То же самое происходит, если копия хромосомы 15, наследуемая от отца, утратила импринтированный участок, несущий в себе отцовскую картину эпигенетических модификаций. В сущности, к синдрому Прадера-Вилли приводит нехватка отцовски-специфической информации. Синдром Ангельмана обусловлен дефектом того же самого участка хромосомы 15, но в данном случае заболевание вызвано нехваткой матерински-специфической информации.
Из всего этого можно сделать далеко идущие выводы. Итак, одна из зон мусорной ДНК (область, контролирующая импринтинг) управляет экспрессией фрагмента мусорной ДНК, который, в свою очередь, кодирует длинную некодирующую РНК. Эта длинная некодирующая РНК, в свою очередь, оказывает определяющее воздействие на регуляцию экспрессии гена, который кодирует целый набор некодирующих РНК. А роль этих некодирующих РНК — в том, чтобы осуществлять регуляцию других РНК, не кодирующих белки. Зная обо всем этом, как-то трудно утверждать, будто мусорная ДНК не обладает никакой функцией.
Синдром Прадера-Вилли и синдром Ангельмана — не единственные заболевания человека, при которых дефекты импринтинга приводят к аномалиям в росте и развитии, а также к ряду других сопутствующих проблем — например, сложностям при обучении. Еще одна взаимосвязанная пара болезней — синдром Сильвера-Рассела24 (проявляется как карликовость) и синдром Беквита-Видемана25 (проявляется как гигантизм). Для некоторых пациентов причиной болезни (той или другой) становятся «родительские» неполадки на одном и том же участке хромосомы 11. Этот импринтинговый локус устроен особенно сложно. Здесь задействовано множество генов и больше одной ОКИ.
Схожие взаимосвязи можно выявить и на других хромосомах. Дети, наследующие обе копии хромосомы 14 от матери, страдают задержкой роста в пренатальный и постнатальный период, однако позже у них развивается ожирение26. Но если обе копии хромосомы 14 ребенок получает от отца, развивается ненормально большая плацента, и дитя появляется на свет с самыми разными проблемами, в том числе с дефектами брюшной стенки27,28.
У большинства этих заболеваний есть столь же редкие разновидности, возникающие из-за эпигенетических погрешностей. Небольшое количество пациентов наследует правильную ДНК от нужного родителя. Эта ДНК не является мутантной. И тем не менее у пациентов возникает импринтинговое заболевание. В этих редких ситуациях обычно нарушаются процессы закрепления и поддержания импринтинга в зиготе и на ранних стадиях развития, что может приводить к неправильному метилированию (или неправильному неметилированию) ОКИ. В результате эта ОКИ отключается или включается тогда, когда не должна этого делать. Вот еще одно подтверждение того, какую важную роль играет общение между мусорной ДНК и эпигенетической аппаратурой.
Назад: Как провести импринтинг заново
Дальше: Влияние катастрофического события

