Книга: Рассказ предка. Паломничество к истокам жизни
Назад: Рандеву № 4 Гиббоны
Дальше: Рандеву № 5 Обезьяны Старого Света
Рассказ Гиббона
На рандеву № 4 мы встречаем крупную группу пилигримов. И теперь могут возникнуть проблемы с установлением родства. (Чем дальше, тем затруднительнее это сделать.) Существует двенадцать видов гиббонов, принадлежащих к четырем основным группам. Это Bunopithecus (группа, представленная одним видом, известным как хулок); настоящие гиббоны Hylobates – шесть видов, самый известный – белорукий гиббон (Hylobates lar); сиаманг (Symphalangus) и номаски (Nomascus) – четыре вида “хохлатых” гиббонов. Сейчас я объясню, как построить схему эволюционных отношений, или филогению, для этих четырех групп.
Генеалогические деревья могут быть укорененными или неукорененными. В случае укорененного древа нам известно, кто является предком. Большинство деревьев в этой книге – укорененные. Неукорененные деревья, напротив, не отражают направление эволюции. Их называют звездчатыми диаграммами. В них не заложена стрела времени, и нельзя сказать, где у них начало, а где конец. Здесь приведены три примера, описывающие отношения четырех родственных групп гиббонов.
Неважно, какая ветвь окажется справа в точке ветвления, а какая – слева. Длина ветвей до сих пор не имела значения (это скоро изменится). Древовидная диаграмма, в которой длина ветвей не несет информации, называется кладограммой (в данном случае неукорененной). Порядок ветвления – вот главная информация, отраженная в кладограмме. Попробуйте перевернуть любую из боковых вилок вокруг центральной горизонтальной линии: это ничего не изменит в схеме отношений между группами.
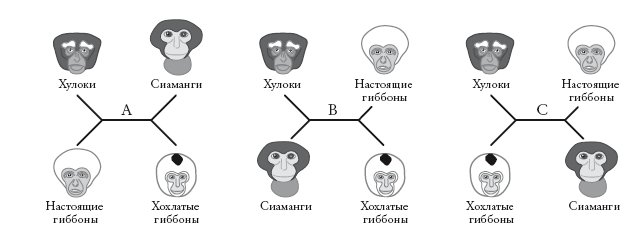
Эти три неукорененные кладограммы описывают возможные отношения четырех видов – при условии, что мы рассматриваем лишь случаи дихотомического ветвления. Как и для укорененных деревьев, случаи разделения на три (трихотомия) и больше ветвей (политомия) мы допускаем, когда у нас недостаточно информации (“неразрешенные” ветви).
Любая неукорененная кладограмма может стать укорененной – для этого нужно указать самую старшую точку на древе (“корень”). Некоторые исследователи – те, на которых мы ссылались при рассмотрении древа в начале этого рассказа, – предлагают для гиббонов укорененную кладограмму слева. Другие предпочитают укорененную кладограмму справа. На первой схеме хохлатые гиббоны (номаски) представлены дальними родственниками всех остальных гиббонов. На второй схеме на их место помещен хулок. Несмотря на это различие, оба древа производны от одного неукорененного дерева (А). Кладограммы отличаются лишь корнем. На первой он расположен на ветви номасков, а на второй – на ветви Bunopithecus.

Как происходит “укоренение”? Самый распространенный способ – расширить древо, включив в него по меньшей мере одну “внешнюю” группу, которая является заведомо далеким родственником по отношению ко всем другим представленным группам. Например, на древе, построенном для гиббонов, внешней группой может быть орангутан или горилла – а еще лучше слон или кенгуру. Можно сколько угодно сомневаться по поводу взаимоотношений групп гиббонов, но мы точно знаем, что общий предок любого гиббона с большими человекообразными обезьянами (или слоном) старше, чем общий предок любого гиббона с любым другим гиббоном. Поэтому, строя древо, включающее гиббонов и крупных человекообразных обезьян, мы не ошибемся, поместив корень где-то между ними.
Легко заметить, что три неукорененных древа, которые я нарисовал, описывают все возможные дихотомические деревья для четырех групп. Для пяти групп будет 15 таких деревьев. Но не стоит и пытаться сосчитать количество возможных деревьев для, скажем, 20 групп. Их сотни миллионов миллионов миллионов. Число резко возрастает с ростом числа групп, которые мы желаем классифицировать, и даже у самого мощного компьютера такие расчеты могут занять целую вечность. Однако в принципе задача довольно проста. Из всех возможных деревьев нужно выбрать те, которые лучше всего объясняют сходства и различия наших групп.
Но что значит – “лучше всего объясняют”? Когда мы рассматриваем выборку животных, количество сходных и отличных черт может оказаться практически бесконечным. Сосчитать их труднее, чем кажется. Нередко один “признак” является неотделимой частью другого. И если мы сочтем эти признаки независимыми, окажется, что на самом деле мы учли одни и те же признаки дважды. Представьте, например, многоножек четырех видов: A, B, C и D. Многоножки A и B сходны во всем, кроме того, что у А конечности красные, а у B – синие. Многоножки C и D сходны друг с другом и отличаются от A и B – но у C конечности красные, а у D – синие. Если мы сочтем цвет конечностей одним “признаком”, мы справедливо поместим A и B в одну группу, а C и D – в другую. Но если мы будем считать каждую ножку из ста отдельным признаком, количество этих признаков перевесит все остальные, и тогда A сгруппируется с C, а B – с D. Очевидно, что в этом случае мы просто сто раз посчитали один и тот же признак. А на самом деле это один признак, потому что цвет всех ста ножек определяется одним эмбриологическим “событием”.
То же верно и для двусторонней симметрии: эмбриогенез таков, что, за редкими исключениями, одна сторона тела животного является зеркальным отражением второй. Ни один зоолог, строя кладограмму, не будет считать дважды “левый” и “правый” признак. Впрочем, не всегда очевидно, какие признаки независимы. Голубю нужна крупная грудина для крепления летательных мышц. А нелетающим птицам, например киви, она не нужна. Должны ли мы считать мощную грудину и способные к полету крылья двумя независимыми признаками, отличающими голубя от киви? Или сочтем их единым признаком на том основании, что состояние одного признака определяет состояние второго – или, по крайней мере, уменьшает его изменчивость? В случае многоножек и зеркальной симметрии правильный ответ очевиден. А в случае грудины – нет. На этот счет может иметься две вполне обоснованные точки зрения.
До сих пор мы говорили о внешнем сходстве и различии. Однако внешние признаки эволюционируют лишь в том случае, если они – проявления последовательностей ДНК. Сегодня мы можем непосредственно сравнить последовательности ДНК. Дополнительное преимущество ДНК заключается в том, что она имеет длинные цепочки, и “текст” ДНК предоставляет гораздо больше признаков, которые можно считать и сравнивать. Проблемы крыльев и грудин просто тонут в огромном потоке данных, которые дает нам ДНК. Более того, многие различия в ДНК “невидимы” для естественного отбора и поэтому являются более “чистыми” свидетельствами родства. Например, многие сочетания нуклеотидов в ДНК синонимичны: они кодируют одну и ту же аминокислоту. Мутация, меняющая сочетание нуклеотидов на синонимичное, невидима для естественного отбора. Однако для генетика такая мутация не хуже любой другой. То же относится и к “псевдогенам” (обычно это случайные копии работающих генов), и ко многим другим “мусорным” последовательностям ДНК, которые располагаются на хромосомах, но не считаются и не используются. Независимая от естественного отбора ДНК получает возможность свободно мутировать, а это обеспечивает специалистов по систематике высокоинформативными данными. Это не отменяет того, что некоторые мутации могут иметь реальный и значительный эффект. Их замечает отбор, они отвечают за видимую глазу красоту и сложность всего живого.
ДНК тоже подвержена проблеме повторного подсчета и нередко представляет собой молекулярный аналог конечностей многоножки. Иногда последовательность представлена многими копиями в разных частях генома. Примерно половина ДНК человека состоит из множественных копий бессмысленных последовательностей, так называемых мобильных элементов, которые, возможно, являются паразитами, захватившими аппарат репликации ДНК, чтобы расселиться по геному. Один из этих паразитических элементов, Alu, у большинства людей представлен более чем миллионом копий. (С ним мы еще встретимся в “Рассказе Ревуна”.) Даже в случае кодирующих участков ДНК гены в некоторых случаях могут быть представлены десятками идентичных (или почти идентичных) копий. Однако на практике повторный подсчет – не такая уж большая проблема: дублированные последовательности ДНК довольно легко обнаружить.
Опасно другое. Иногда обширные области ДНК проявляют таинственное сходство с последовательностями ДНК отдаленных видов. Никто не сомневается, что птицы ближе к черепахам, ящерицам, змеям и крокодилам, чем к млекопитающим (рандеву № 16). Однако последовательности ДНК птиц и млекопитающих имеют большее сходство, чем можно ожидать. И у тех, и у других в некодирующей ДНК наблюдается избыток пар Г – Ц. Пары Г – Ц химически стабильнее пар A – T. Возможно, теплокровные виды (птицы и млекопитающие) нуждаются в более “крепкой” ДНК. Каково бы ни было объяснение, мы должны быть осторожны и не позволять этому смещению Г – Ц убедить нас в том, что все теплокровные животные – близкие родственники. Хотя специалисты по систематике утверждают, что ДНК – это все, о чем можно мечтать, нельзя забывать: мы по-прежнему многого не понимаем в геноме.
Как использовать информацию, заключенную в ДНК? Литературоведы, изучая происхождение текстов, используют ту же технику, что и эволюционные биологи. И, хотя это звучит неправдоподобно, одним из лучших примеров является проект по изучению “Кентерберийских рассказов”. Участники этого международного проекта использовали инструменты эволюционной биологии, чтобы проследить историю 85 списков “Кентерберийских рассказов”. Эти манускрипты – наша главная надежда на восстановление утраченного оригинала. Как и ДНК, текст Чосера уцелел благодаря многократному копированию. При этом каждый раз при копировании возникали случайные изменения. Тщательно оценив накопленные отличия, исследователи реконструируют историю копирования и строят эволюционное древо – потому что это настоящий эволюционный процесс, при котором с каждым поколением накапливаются ошибки. Способы реконструкции эволюции ДНК и текста настолько похожи, что каждый из них может служить иллюстрацией другого.
Отвлечемся от гиббонов и займемся Чосером, а именно четырьмя из 85 списков “Кентерберийских рассказов”. Эти рукописи называются: “Британская библиотека” (British Library), “Крайст-Черч” (Christ Church), “Эджертон” (Edgerton) и “Хенгурт” (Hengwrt). Вот две первые строки “Общего пролога”:
Когда Апрель обильными дождями
Разрыхлил землю, взрытую ростками…
Теперь сравним. Список из Британской библиотеки гласит:
Whan that Aprylle / wyth hys showres soote
The drowhte of Marche / hath pcede to the rote
“Крайст-Черч”:
Whan that Auerell wt his shoures soote
The droght of Marche hath pced to the roote
“Эджертон”:
Whan that Aprille with his showres soote
The drowte of marche hath pced to the roote
“Хенгурт”:
Whan that Aueryll wt his shoures soote
The droghte of March / hath pced to the roote
Первое, что нужно сделать с последовательностью ДНК или текстом, – выявить сходства и различия. Для этого нужно их “выровнять” – а это бывает не так-то просто: тексты могут быть фрагментарными и иметь разную длину. Здесь очень помогает компьютер, но чтобы выровнять первые две строки “Общего пролога”, он не понадобится. На рисунке выделены 14 позиций, по которым тексты не совпадают.
Вторая и пятая позиции представлены даже не двумя вариантами, а тремя. В целом это дает 16 “различий”. После того, как мы составили список различий, нужно определить, какое древо лучше всего их объясняет. Есть множество способов это сделать, и все их можно применить и к животным, и к текстам. Самый простой пример – группировка текстов на основе общего сходства. Как правило, при этом используют варианты следующего метода. Сначала мы находим пару наиболее сходных текстов. Затем мы используем эту пару в качестве единого усредненного текста и сравниваем его с оставшимися, чтобы найти следующую пару наиболее сходных текстов. Так мы последовательно формируем новые пары, пока не получится генеалогическая схема. Такой способ построения деревьев используется чаще всего и называется методом поиска ближайшего соседа (neighbourpmmg). Он прост, но не учитывает логику эволюционного процесса: мы просто оцениваем сходство. Поэтому сторонники “кла-дистического” подхода в систематике (он основан на принципах эволюции) предпочитают иные методы. Первым был разработан метод парсимонии (экономии).
Экономия, как мы узнали из “Рассказа Орангутана”, означает здесь экономичность объяснения. В эволюции (животного ли, манускрипта ли) самым экономичным является объяснение, подразумевающее наименьшее число эволюционных изменений. Если два текста объединены общим признаком, самое экономичное объяснение будет гласить: оба текста унаследовали этот признак от общего предка. Конечно, и у этого правила есть исключения, однако чаще всего оно верно. Метод парсимонии – по крайней мере в теории – сравнивает все возможные деревья и выбирает то, в котором количество изменений минимально.
Когда мы сравниваем деревья по их экономичности, некоторые виды признаков оказываются бесполезными. Признаки, уникальные для манускрипта или вида животного, неинформативны. В методе поиска ближайшего соседа такие признаки учитываются, однако метод парсимонии целиком их игнорирует. Метод парсимонии опирается на информативные признаки, то есть такие, которые наблюдаются более чем в одном манускрипте. Предпочтительным древом является объясняющее максимальное количество информативных признаков общим происхождением. В строках Чосера пять таких информативных признаков. Четыре из них делят манускрипты на следующие группы:

{“Британская библиотека” + “Эджертон”} и (“Крайст-Черч” + “Хенгурт”}
Эти признаки выделены первой, третьей, седьмой и восьмой вертикальными линиями. Пятый признак – косая черта – выделен двенадцатой вертикальной линией. По этому признаку манускрипты подразделяются на другие группы:
{“Британская библиотека” + “Хенгурт”} и {“Крайст-Черч” + “Эджертон”}
Полученные результаты противоречат друг другу. Мы не можем построить древо, в котором каждое изменение отображалось бы лишь один раз. Самым приемлемым окажется древо, изображенное ниже (заметьте – оно неукорененное). Эта схема сокращает противоречия до минимума: мы повторно учитываем лишь один признак – косую черту.
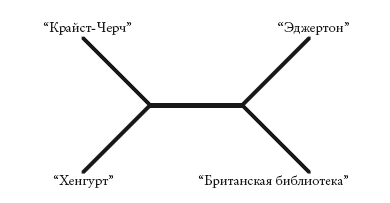
Вообще-то я не уверен, что мы сделали правильное предположение. В текстах часто встречаются совпадения и реверсии, особенно если смысл строк при этом не меняется. Средневековый переписчик наверняка не испытывал угрызений совести, изменяя написание, и еще меньше его волновали вставки или удаления знаков, например косой черты. В этом случае информативнее такие изменения, как перестановка слов. В генетике аналогами таких изменений являются “редкие геномные изменения”: крупные вставки, делеции и дупликации ДНК. Мы можем оценить информативность, присвоив большее или меньшее значение (вес) различным типам признаков. Недостоверные или слишком частые изменения при подсчете будут иметь меньший вес. А редкие изменения, которые служат надежными показателями родства, – больший вес. Повышенный вес признака говорит о том, что мы не хотим учитывать его дважды. Таким образом, наиболее экономное древо – то, которое имеет наименьший общий вес.
Метод парсимонии широко используется для поиска эволюционных деревьев. Но в том случае, когда конвергенций и реверсий слишком много – а это случается и с последовательностями ДНК, и с текстами Чосера, – метод парсимонии может оказаться недостоверным. Эта проблема известна как “эффект притяжения длинных ветвей”.
Кладограммы – как укорененные, так и неукорененные – отражают лишь порядок ветвления. Филограммы, или филогенетические деревья, похожи на кладограммы, но в них длина ветвей несет дополнительную информацию. Обычно длина ветвей отражает эволюционное расстояние: длинные ветви обозначают крупные изменения, а короткие – мелкие. На основе первой строки “Кентерберийских рассказов” можно построить следующую филограмму.
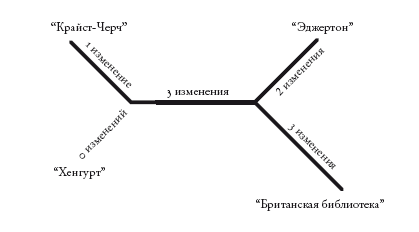
Здесь длина ветвей не слишком различается. Но представьте, что будет, если два манускрипта сильно отличаются от двух других. Тогда ветви первых манускриптов будут очень длинными. Однако изменения могут оказаться не уникальными. Изменения могут случайно оказаться идентичными изменениям в другом месте древа. Но с наибольшей вероятностью (именно в этом заключается проблема) они совпадут с изменениями на другой длинной ветви. Ведь длинные ветви – это те, в которых произошло наибольшее число изменений. И если изменений окажется слишком много, две длинные ветви на филограмме будут отображаться как родственные, даже если это не так. Таким образом, метод парсимонии, основываясь на простом подсчете изменений, может ошибочно сгруппировать две самые длинные ветви, “притянуть” их друг к другу.
Эффект притяжения длинных ветвей – серьезная помеха для систематики. Он проявляется везде, где много конвергенций и реверсий. К сожалению, эту проблему нельзя решить простым увеличением объема рассматриваемого текста. Наоборот, чем больше текст, тем выше вероятность обнаружения случайных совпадений. Про такие деревья говорят, что они лежат в “зоне Фельзенстайна” (звучит устрашающе!), названной в честь американского биолога Джо Фельзенстайна. Увы, ДНК особенно подвержена эффекту притяжения длинных ветвей. Основная причина в том, что в ДНК всего четыре “буквы”. Поскольку большинство изменений затрагивают всего одну “букву”, случайные мутации с высокой вероятностью могут привести к совпадениям. Так возникает притяжение длинных ветвей. Очевидно, что для таких случаев нужна альтернатива методу парсимонии. Она существует – это метод правдоподобия. В последнее время он используется все чаще.
Оценка правдоподобия требует больше вычислительных мощностей, чем метод парсимонии, поскольку здесь мы учитываем длину ветвей. Таким образом, приходится иметь дело с еще большим количеством деревьев: вдобавок к рассмотрению возможных схем ветвления мы должны учитывать возможные длины ветвей. Геркулесов труд! Поэтому, несмотря на упрощенные методы вычисления, компьютеры пока могут подвергнуть анализу небольшое количество видов.
Термин “правдоподобие” здесь имеет вполне точное значение. Возьмем древо определенной формы (с учетом длины ветвей). Из всех возможных эволюционных траекторий, посредством которых может сформироваться филогенетическое древо данной формы, всего несколько могут привести к тому тексту, который мы сейчас видим. "Правдоподобие” данного древа – это ничтожно малая вероятность получения реально существующих текстов, а не каких-нибудь текстов, которые могут появиться на таком древе. Величина правдоподобия для древа очень мала, однако это не мешает сравнить одну малую величину с другой, чтобы выбрать нужную.

Неукорененное филогенетическое древо первых 250 строк 24 списков «Кентерберийских рассказов». Здесь представлен набор списков, изученный в рамках проекта «Кентерберийские рассказы». Сокращения соответствуют тем, что использованы в проекте. Схема построена методом парсимонии, на каждой ветви указаны индексы бутстреп-поддержки. Для четырех списков, которые обсуждаются нами, указаны их полные названия.
Выбирать "лучшее” древо методом правдоподобия можно по-разному. Самый простой способ – искать наиболее правдоподобное древо. Это метод максимального правдоподобия. Однако то, что это наиболее правдоподобное древо, вовсе не означает, что другие деревья не окажутся почти столь же правдоподобными. Совсем недавно было предложено не искать одно самое правдоподобное древо, а рассматривать все возможные. При этом степень "доверия” к древу должна зависеть от его правдоподобия. Этот подход представляет собой альтернативу методу правдоподобия и известен как байесовский метод. Если схема ветвления подтверждается большим количеством правдоподобных деревьев, мы заключаем, что эта схема с высокой вероятностью верна. Конечно, как и в методе максимального правдоподобия, мы не можем проверить все деревья. Но существуют способы упрощения вычислений, и они довольно неплохо работают.
Степень нашего доверия древу, которое мы в итоге выберем, зависит от того, насколько мы уверены в правильности каждого разветвления. Поэтому возле точек ветвления часто указывают степень “уверенности” в них. При использовании байесовского метода правдоподобие точек ветвления вычисляется автоматически, однако для других методов, таких как парсимония или максимальное правдоподобие, необходимы альтернативные способы подсчета. Чаще всего используют метод бутстрепа: многократно обсчитываются выборки данных, и оценки сравниваются с результатами для всего древа. Так мы можем понять, насколько древо устойчиво к ошибкам. Чем больше индекс бутстреп-поддержки, тем надежнее точка ветвления. Правда, точно интерпретировать полученные индексы бывает непросто. По сходному алгоритму работают методы “складного ножа” (jackknife) и “поддержки Бремера”. Все они служат для оценки достоверности точек ветвления.
Прежде чем оставить литературу, рассмотрим итоговое древо, построенное для первых 250 строк в 24 манускриптах Чосера. Это филограмма, на которой информативна не только схема ветвления, но и длина ветвей. На схеме видно, какие списки почти идентичны, а какие сильно отличаются от остальных. Эта филограмма неукорененная, то есть не указывает на то, какой из 24 манускриптов ближе всех к “оригиналу”.
Вернемся к гиббонам. Принцип парсимонии предполагает существование четырех групп. Ниже приведена укорененная диаграмма, основанная на морфологических признаках. Здесь виды рода Hylobates (настоящие гиббоны) группируются вместе, как и виды рода Nomascus. Обе группы поддерживаются высокими индексами бутстреп-поддержки (указаны над ветвями). Однако в нескольких местах порядок ветвлений не определен. Хотя Hylobates и Bunopithecus вроде бы формируют группу, индекс бутстреп-поддержки (63) представляется неубедительным для тех, кто умеет читать подобные руны. Морфологических признаков для построения древа недостаточно.
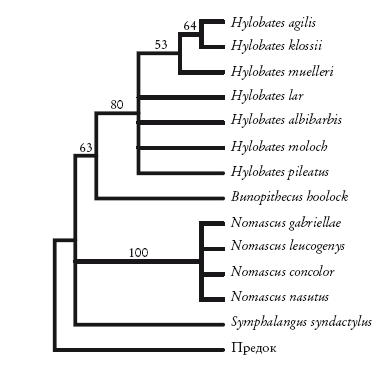
Укорененная кладограмма гиббонов, построенная на основе морфологии. Geissmann [100].
По этой причине Кристиан Роос и Томас Гайсман, ученые из Германии, обратились к молекулярной генетике, а именно к участку митохондриальной ДНК, который называют контрольным регионом. Взяв ДНК шести гиббонов, они расшифровали последовательности, выровняли их и провели анализ с помощью методов поиска ближайшего соседа, парсимонии и максимального правдоподобия. Самый убедительный результат был получен с помощью метода максимального правдоподобия, который лучше других методов справляется с эффектом притяжения длинных ветвей. Итоговое древо, где показаны отношения между четырьмя группами, приведено здесь. Значения бутстреп-поддержки на этом древе вполне убедительны. Так что, на мой взгляд, это то, что нам нужно.
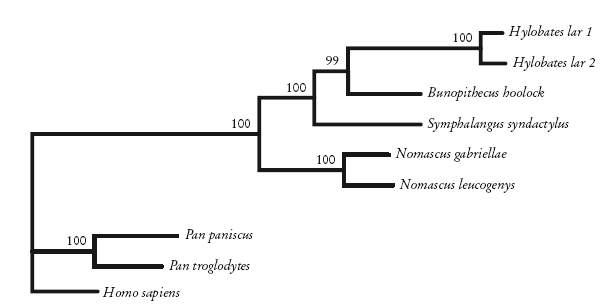
Кладограмма гиббонов, построенная с помощью метода максимального правдоподобия по данным ДНК. Roos and Geissmann [246].
Видообразование у гиббонов произошло сравнительно недавно. Однако если изучать все более удаленные виды, которые будут разделены все более длинными ветвями, в конце концов даже изощренные методы Байеса и максимального правдоподобия откажутся нам служить. В определенный момент недопустимо большая доля сходств окажется случайной. Когда это происходит с ДНК, говорят, что наступило насыщение. И тогда ни один метод не поможет реконструировать схему родственных отношений: действие времени заглушает “филогенетический” сигнал. Особенно остро этот вопрос стоит в отношении нейтральных мутаций ДНК. Давление естественного отбора не позволяет генам сбиваться с пути, удерживая их в узком диапазоне. В некоторых случаях самые важные функциональные гены могут оставаться практически неизменными сотни миллионов лет. Однако для псевдогена, с которого никогда ничего не считывается, таких промежутков времени достаточно для безнадежно сильного насыщения. В таких случаях нам приходится искать другие данные. Одна из самых перспективных идей – использование редких геномных мутаций, о которых я упоминал. Эти изменения затрагивают значительные участки ДНК, а не одну “букву”. Поскольку такие перестройки редки и, как правило, уникальны, проблема случайного сходства не возникает. Эти мутации могут выявлять неожиданные родственные связи. Мы убедимся в этом, когда к толпе пилигримов присоединятся гиппопотамы. (Вот увидите, они расскажут удивительные вещи!)
А теперь обобщим то, что узнали из “Рассказа Митохондриальной Евы” и “Рассказа Неандертальца”. Cчитается, что для группы видов должно существовать лишь одно эволюционное древо. Однако из “Рассказа Митохондриальной Евы” видно, что на основе разных участков ДНК (а также для разных признаков или разных частей тела) можно построить разные деревья. Мне кажется, эта проблема заложена в самой идее филогенетических деревьев видов. Ведь вид представляет собой сложную мозаику фрагментов ДНК, полученных из разных источников. Мы увидели, что каждый ген, да и каждая “буква” ДНК, эволюционирует независимо. Для каждого фрагмента ДНК и каждого признака организма можно построить свое эволюционное древо.
С доказательствами этого мы сталкиваемся каждый день – и поэтому их не замечаем. Если предьявить марсианину гениталии мужчины, женщины и самца гиббона, пришелец, не колеблясь, решит, что наиболее близким родством связаны два самца. И правда: ген, определяющий мужской пол (SRY), никогда не бывал в теле женщины – а если и бывал, то задолго до того, как мы разошлись с гиббонами. Морфологи традиционно делают исключение для половых признаков, избегая “бессмысленных” классификаций. Однако такого рода проблемы встречаются на каждом шагу. Мы столкнулись с этим в “Рассказе Митохондриальной Евы”, когда говорили о группе крови ABo. Если рассматривать гены группы крови, окажется, что мой ген группы крови B сближает меня с шимпанзе с группой крови B, а не A. Все это касается не только генов, определяющих пол, или генов группы крови. Нет, при определенных обстоятельствах эта проблема затрагивает абсолютно все гены и признаки. Большинство молекулярных и морфологических признаков указывает на то, что шимпанзе – наш ближайший родственник. Однако меньшая доля признаков указывает на то, что наш ближайший родственник – горилла, или что шимпанзе ближе всего к гориллам, а не к человеку.
Не удивляйтесь! Популяция, предковая для всех трех видов, должна быть очень изменчивой, и у каждого гена в популяции должно быть несколько вариантов. Каждый из вариантов передается по своей линии. Вполне возможно, например, что человек и горилла получили некий ген от одной линии, а шимпанзе – от другой. После этого нужно только, чтобы разошедшиеся в древности генетические линии тянулись непрерывно до точки расхождения человека и шимпанзе. И получится, что человек произошел от одной линии, а шимпанзе – от другой.
Приходится признать, что одно древо не описывает весь эволюционный сюжет. Ничто не мешает нам продолжать строить деревья для видов, однако нужно помнить, что эти деревья представляют не более чем обобщение множества генных деревьев. Интерпретировать деревья можно двумя способами. Первый – традиционная генеалогическая интерпретация. Один вид является ближайшим родственником другого, если из всех рассмотренных видов именно с ним его связывает самый поздний общий предок. Второй способ интерпретации, мне кажется, только предстоит освоить. Согласно этому подходу, построенное для группы видов древо отражает родственные отношения большей части генов. То есть древо показывает результаты, за которые гены высказались “большинством голосов”.
Мне больше нравится идея голосования генов. Поэтому, когда я говорю о родстве видов, его нужно понимать именно так. Все филогенетические деревья, которые я здесь обсуждаю – касаются ли они животных, растений, грибов или бактерий, – нужно рассматривать как схемы, отражающие идеи “генного большинства”.
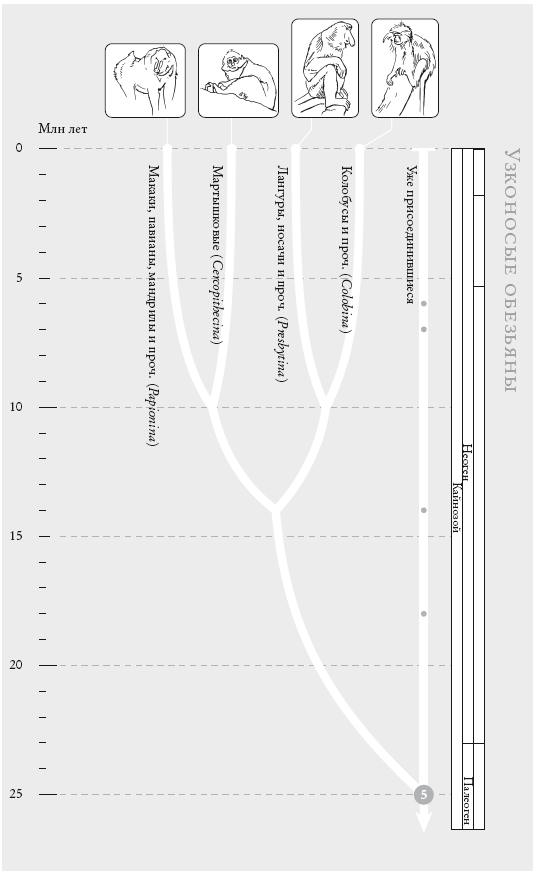
Узконосые обезьяны. Это общепризнанное филогенетическое древо, построенное примерно для ста видов обезьян Старого Света. (Кружки на концах ветвей указывают на количество видов в каждой группе: отсутствие кружка означает 1–9 известных видов, небольшой кружок соответствует 10–99 видам, круг побольше – 100–999 и т. д. Каждая из представленных здесь четырех групп объединяет 10–99 видов.)
На рис. (слева направо): мандрил (Mandrillus sphinx), краснохвостая мартышка (Cercopithecus ascanius), носач (Nasalis larvatus), ангольский чернобелый колобус (Colobus angolensis).
Назад: Рандеву № 4 Гиббоны
Дальше: Рандеву № 5 Обезьяны Старого Света

